Download
1 / 1
10 likes | 248 Views
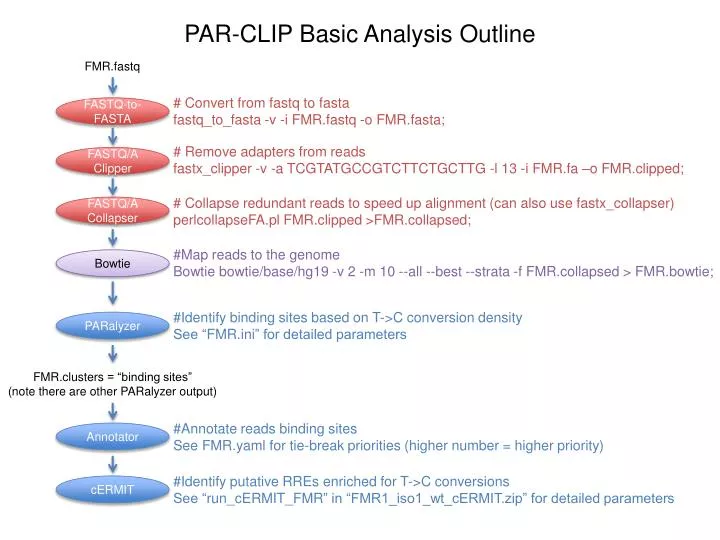
PAR-CLIP Basic Analysis Outline. FMR.fastq. # Convert from fastq to fasta fastq_to_fasta -v - i FMR.fastq -o FMR.fasta ;. FASTQ-to-FASTA. # Remove adapters from reads fastx_clipper -v -a TCGTATGCCGTCTTCTGCTTG -l 13 - i FMR.fa –o FMR.clipped ;. FASTQ/A Clipper.

E N D
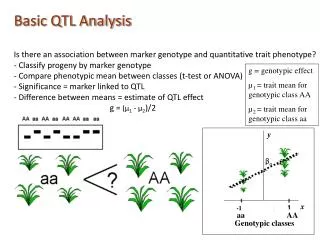
PAR-CLIP Basic Analysis Outline FMR.fastq • # Convert from fastq to fasta • fastq_to_fasta -v -iFMR.fastq -o FMR.fasta; FASTQ-to-FASTA • # Remove adapters from reads • fastx_clipper -v -a TCGTATGCCGTCTTCTGCTTG -l 13 -iFMR.fa–o FMR.clipped; FASTQ/A Clipper # Collapse redundant reads to speed up alignment (can also use fastx_collapser) perlcollapseFA.plFMR.clipped >FMR.collapsed; FASTQ/A Collapser #Map reads to the genome Bowtie bowtie/base/hg19 -v 2 -m 10 --all --best --strata -f FMR.collapsed > FMR.bowtie; Bowtie #Identify binding sites based on T->C conversion density See “FMR.ini” for detailed parameters PARalyzer FMR.clusters = “binding sites” (note there are other PARalyzer output) #Annotate reads binding sites See FMR.yaml for tie-break priorities (higher number = higher priority) Annotator #Identify putative RREs enriched for T->C conversions See “run_cERMIT_FMR” in “FMR1_iso1_wt_cERMIT.zip” for detailed parameters cERMIT