Download

1 / 10
130 likes | 395 Views
miRNA. Published online before print March 7, 2006, 10.1073/pnas.0601268103 PNAS | March 14, 2006 | vol. 103 | no. 11 | 3951-3952 Institution: Bolivia: PNAS Sponsored Commentary: MicroRNAs: New players in an old game Malavika Gupta, and Gary Brewer*

E N D
miRNA Published online before print March 7, 2006, 10.1073/pnas.0601268103 PNAS | March 14, 2006 | vol. 103 | no. 11 | 3951-3952Institution: Bolivia: PNAS Sponsored Commentary: MicroRNAs: New players in an old game Malavika Gupta, and Gary Brewer* Department of Molecular Genetics, Microbiology, and Immunology, University of Medicine and Dentistry of New Jersey–Robert Wood Johnson Medical School, Piscataway NJ 08854 One of the cardinal steps in regulating gene expression is mRNA decay, and the numerous pathways and mechanisms that exist to regulate it underscore its importance. mRNA decay is regulated by trans-acting factors that assemble on cis-acting elements (1, 2). Together, they serve to up- or down-regulate a given mRNA. Some of the mechanisms that regulate mRNA levels involve surveillance pathways such as nonsense-mediated decay (NMD) and nonstop decay (NSD). The NMD (nonsense-mediated decay ) pathway limits accumulation of mRNAs that contain a premature termination codon and whose translation would produce a truncated protein. In NSD (nonstop decay ), mRNAs that do not contain a termination codon because of improper poly(A) site selection within the coding region are rapidly degraded by the exosome, a complex of 3'->5' exoribonucleases (3). Other pathways involve recognition of 3' UTR sequences by specific RNA-binding proteins. For example, in AU-rich element (ARE)-mediated mRNA decay (AMD), binding of specific ARE recognition proteins to the 3' UTR initiates mRNA degradation (1, 4).
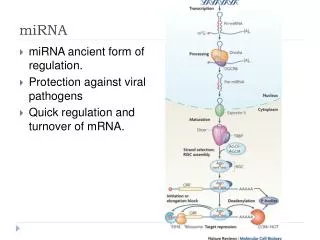
To one degree or another, all these decay pathways involve the step-wise deconstruction of a mRNA involving 3'->5' trimming of the poly(A) tail, a process referred to as deadenylation; this is followed by removal of the 5' m7GpppG cap and both 5'->3' and 3'->5' degradation of the mRNA body (5–7). This step-wise mechanism, first elucidated in Saccharomyces cerevisiae, has been recognized for some time now. Another mRNA decay pathway that has garnered much attention lately is RNA interference (RNAi). First discovered in Caenorhabditis elegans (8), RNAi has now been observed in several other multicellular organisms, including mammals. RNAi is triggered either by a small interfering RNA (siRNA) or, in some cases, by a microRNA (miRNA) that induces mRNA degradation via endoribonucleolytic cleavage within the site of si/miRNA–mRNA annealing. siRNAs derive from sources such as double-stranded RNA, transposons, and viruses and are perfectly complementary to their mRNA targets (9–11). miRNAs are {approx}22 nt in length and are encoded within the genomes of both plants and animals. miRNAs contain regions possessing imperfect complementarity to 3' UTRs of mRNA subsets to which they anneal. This leads to translational silencing to posttranscriptionally control gene expression (12). In this issue of PNAS, Wu et al. (13) demonstrate that a miRNA can also promote rapid mRNA degradation by accelerating the initial rate-limiting step, deadenylation.
The life of a miRNA begins as a miRNA precursor called primary miRNA (pri-miRNA). In metazoans, the enzyme Drosha catalyzes the first cleavage event that results in the production of a pre-miRNA intermediate. With the assistance of Exportin5, this intermediate travels to the cytoplasm, where it undergoes further cleavage by a second enzyme called Dicer. Cleavage by Dicer leaves an RNA duplex that is unwound, and the so-called guide strand, which contains complementarity to mRNA targets, assembles with Argonaute proteins and others to form the RNA-induced silencing complex (RISC) (14). A large body of evidence indicates miRNAs to be translational repressors and siRNAs to be purveyors of mRNA degradation. A few exceptions to translational silencing by miRNAs have begun trickling into the literature, however. For example, mammalian miR196a is perfectly complementary to one of its target transcripts, HOXB8, except for a single G:U wobble base pair (Fig. 1) (11). miR196a directs endoribonucleolytic cleavage of HOXB8 mRNA, which encodes one member of a group of related transcription factors involved in animal development.
Although miR196a exhibits near-perfect complementarity to its target mRNA, miR125b and the miRNA known as let-7, do not (Fig. 1) (15). Although Wu and Belasco (15) demonstrated that miR125b target association did repress translation, this miRNA surprisingly led to reduced mRNA levels as well. In work presented here, they demonstrate that miR125b and let-7 increase mRNA decay rates upon association with their target mRNAs, not by endoribonucleolytic cleavage but rather by promoting rapid deadenylation.
transcriptional pausing Institution: Bolivia: PNAS Sponsored Published online before print March 13, 2006, 10.1073/pnas.0600508103PNAS March 21, 2006 | vol. 103 | no. 12 | 4439-444 biologicla science/biophysics: Thermodynamic and kinetic modeling of transcriptional pausing Vasisht R. Tadigotla, Dáibhid Ó Maoiléidigh, Anirvan M. Sengupta, Vitaly Epshtein, Richard H. Ebright, Evgeny Nudler, and Andrei E. Ruckenstein The basis for our quantitative analysis is the structural and mechanistic model of the elongation complex (EC), sketched in Fig. 1. The EC consists of a melted DNA duplex region of 12–14 nt (transcription bubble) enclosed within RNAP and stabilized by interactions with the enzyme and with the last 8 or 9 nt of the synthesized RNA transcript (the DNA–RNA hybrid). The RNA transcript upstream of the hybrid exits RNAP via the "RNA exit channel"; whereas the duplex DNA downstream of the bubble threads through a "sliding clamp” in the enzyme, which holds on tightly to the DNA while allowing for smooth sliding during transcription elongation.
The "secondary channel" provides the access of the incoming nucleoside triphosphate (NTP) to the active center where the catalysis of phosphodiester bond formation takes place, resulting in the elongation of the RNA transcript by one nucleotide. Immediately after the transcript elongation step the EC is in the so-called "pretranslocated" state (translocational state 0) in which the 3' end of the transcript overlaps the catalytic site. The next incorporation step requires that RNAP translocate forward by one base pair, into the "posttranslocated" state (translocational state +1), making the catalytic center available for the binding of the next NTP.
Genomes of most eukaryotes are populated by DNA copies of parasitic elements transposons Institution: Bolivia: PNAS Sponsored Published online before print March 14, 2006, March 21, 2006 | vol. 103 | no. 12 | 4540-4545 biological science/genetics Self-synthesizing DNA transposons in eukaryotes Vladimir V. Kapitonov*, and Jerzy Jurka* Genomes of most eukaryotes are populated by DNA copies of parasitic elements known as transposable elements (TEs) capable of reproducing themselves in the host genome in a non-Mendelian fashion (1, 2). Understanding the biology of transposable elements is of great importance because of their increasingly well documented impact on the host genome (2, 3). Moreover, transposable elements can be used as powerful tools in genetic engineering (4). Despite an enormous diversity of eukaryotic TEs, they belong to only two types, called retrotransposons and DNA transposons. Whereas a retrotransposon is transposed (retroposed) via reverse transcription of its mRNAs, a DNA transposon is transposed via transfer of its genomic copy from one site to another.
Each type includes different classes and families of TEs composed of autonomous and nonautonomous elements. Whereas an autonomous element encodes a complete set of enzymes characteristic of its family, a nonautonomous element encodes none, or only some of them, and depends on enzymes encoded by its autonomous relative. Transposition of a retrotransposon is catalyzed by reverse transcriptase and endonuclease (EN) domains of a polyprotein encoded by itself or by other retrotransposons. All retrotransposons can be further divided into two subclasses called LTR and non-LTR retrotransposons (5). In addition to the reverse transcriptase/EN polyprotein, most non-LTR retrotransposons code for a second protein characterized by poorly understood activities, including RNA/DNA binding, chaperone, and esterase. An mRNA molecule expressed during transcription of the genomic non-LTR retrotransposon is reverse transcribed and inserted in the genome (5). LTR retrotransposons, including endogenous retroviruses, represent the most complex TEs in eukaryotes. An LTR retrotransposon may carry three ORFs coding for the gag, env, and pol proteins, the latter is composed of the reverse transcriptase, EN, and aspartyl protease domains (5). The endonuclease domain in LTR retrotransposons is usually called integrase (INT) and is distantly related to the DDE transposases (TPase) encoded by Mariner DNA transposon