Download
1 / 27
270 likes | 283 Views
Проблемы нахождения ортологов. BranchClust - филогенетический алгоритм отбора семейств генов. Отбор семейств генов на примере 5 геномов протеобактерий. Бесплатный SCP-клиент. BranchClust - филогенетический алгоритм отбора семейств генов. Семейство АТФ-синтаз. Случай 2-х бактерий и 2-х архей.

E N D
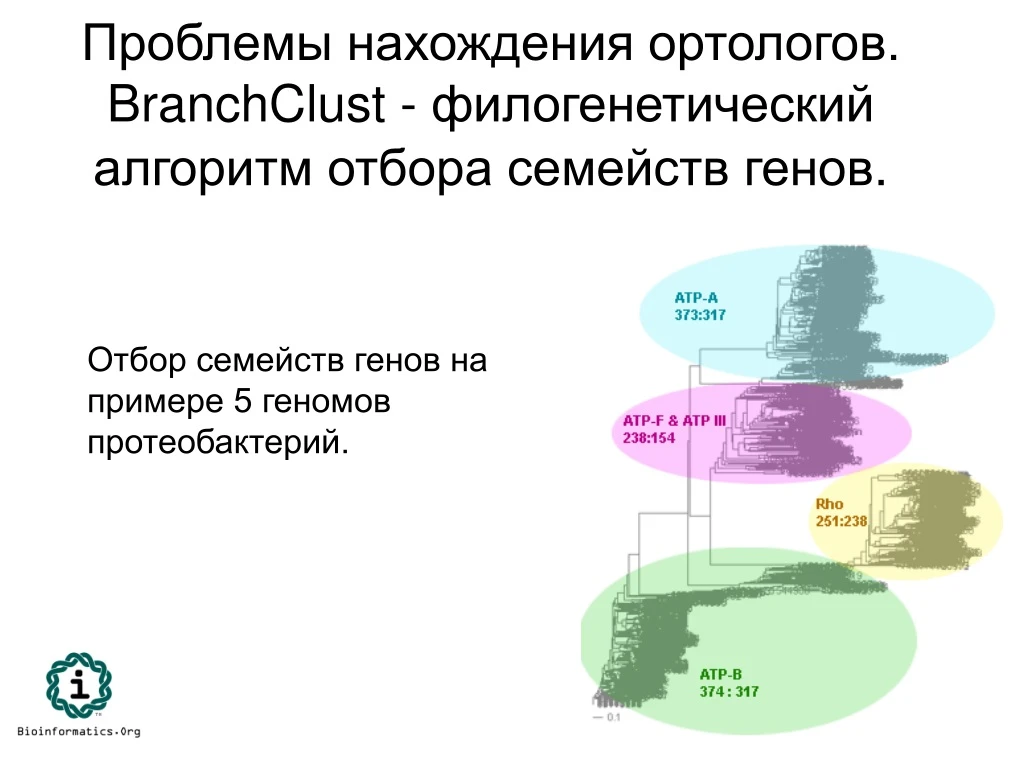
Проблемы нахождения ортологов. BranchClust - филогенетический алгоритм отбора семейств генов. Отбор семейств генов на примере 5 геномов протеобактерий.
BranchClust - филогенетический алгоритм отбора семейств генов
Семейство АТФ-синтаз Случай 2-х бактерий и 2-х архей ATP-B (non-catalytic subunit) ATP-A (catalytic subunit) ATP-A ATP-A Escherichia coli Escherichia coli ATP-B Methanosarcina mazei ATP-B Methanosarcina mazei ATP-F ATP-A ATP-A ATP-A ATP-A ATP-B ATP-B ATP-B ATP-B Sulfolobus solfataricus Sulfolobus solfataricus ATP-A ATP-A ATP-B ATP-B Bacillus subtilis Bacillus subtilis ATP-F Метод RBH не отбирает ни ATP-A, ни ATB-B
Families of ATP-synthases Phylogenetic Tree Familyof ATP-A Sulfolobus solfataricus ATP-A Methanosarcina mazei Bacillus subtilis ATP-A ATP-A ATP-A Escherichia coli ATP-F Bacillus subtilis ATP-B Escherichia coli Escherichia coli ATP-F ATP-B Bacillus subtilis ATP-B ATP-B Sulfolobus solfataricus Methanosarcina mazei Family of ATP-F Family of ATP-B
BranchClust Algorithm genome 1 genome 2 BLAST genome 3 hits genome i genome N superfamily tree dataset of N genomes www.bioinformatics.org/branchclust
BranchClust Algorithm www.bioinformatics.org/branchclust
BranchClust Algorithm Root positions Superfamily of DNA-binding protein Superfamily of penicillin-binding protein 13 gamma proteo bacteria 13 gamma proteo bacteria www.bioinformatics.org/branchclust
Пример: 5 протеобактерий 1 Gamma-proteobacteria Escherichia_coli_K_12_substr__MG1655_uid57779 2 Beta-proteobacteria Bordetella_parapertussis_12822_uid57615 3 Alpha-proteobacteria Rickettsia_prowazekii_Madrid_E_uid61565 4 Epsilon-proteobacteria Helicobacter_pylori_26695_uid57787 5 Delta-proteobacteria Desulfovibrio_vulgaris_DP4_uid58679
Дерево построено конкатенацией ортологов из 31 семейства из 191 видов Ciccarelli et al. 2006, Science
Редактируем файл .bash_profile >cd >vi .bash_profile export PATH=/usr/local/biotools/bin:/usr/local/biotools/data/:$PATH export BLASTMAT=/usr/local/biotools/data/ Устанавливаем BioPerl Копируем пакет bioperl в домашнюю директорию
Создать один файл, содержащий все 5 геномов >perl create_one_faa.pl #!usr/bin/perl -w #create dir if (!opendir(DIR,"")){ mkdir("fasta_all"); }else{ close(DIR); } system(" > fasta_all/allgenomes.faa"); while(defined($file=glob("fasta/*.faa"))) { system("cat $file >> fasta_all/allgenomes.faa"); }
Форматировать файл для использования blast >perl format_faa.pl #! /usr/bin/perl -w system("formatdb -o T -p T -i fasta_all/allgenomes.faa"); bio568b-2:test mariap$ ls fasta_all allgenomes.faa allgenomes.faa.pin allgenomes.faa.pni allgenomes.faa.psi allgenomes.faa.phr allgenomes.faa.pnd allgenomes.faa.psd allgenomes.faa.psq
Запускаем программу BLAST Примерное время работы для нашего файла - 30 минутт (когда запущен один вариант программы) >perl do_blast.pl #!/usr/bin/perl -w #create 'blast' dir if it doesn't exist if (!opendir(DIR,"blast")){ mkdir("blast"); }else{ close(DIR); } $blast_input="fasta_all/allgenomes.faa"; $blast_output="blast/all_vs_all.out"; #system("blastall -i $blast_input -d $blast_input -p blastp -o $blast_output -I T -e 1E-4 -F F -W 2 -m 8"); system("blastall -i $blast_input -d $blast_input -p blastp -o $blast_output -I T -e 1E-4 -m 8");
Отслеживаем свои процессы >perl do_blast.pl - убить ctrl-c >perl do_blast.pl & (запуск на background) Просмотр процессов >ps >ps aux >top Убить свои процессы >kill -9 pid где pid - номер процесса
Результаты blast > more blast/all_vs_all.out | wc -l 221973 more blast/all_vs_all.out
Обрабатываем результаты blast my $in = new Bio::SearchIO(-format => 'blasttable', -file => "$infile"); # Because it is blast of a database of N genomes against itself, # first hit for each gene is the gene itself. # That is why we assemble only hits. while(my $result = $in->next_result){ while($hit = $result->next_hit()){ # take only first hsp for every hit, it has the best e-value # exctract gene number $hit->name()=~/\|(.+?)\|/s; $gene=$1; print OUT "$gene\t"; } print OUT "\n"; } close (OUT); > more blast/all_vs_all.out | wc -l 221973 > perl parse_blast.pl #! /usr/bin/perl -w use lib "/Users/mariap/bioperl-1.5-my"; use Bio::SearchIO; #create 'parsed' if it doesn't exist if(!opendir(DIR,"parsed")){ mkdir("parsed"); }else{ close(DIR); } $infile="blast/all_vs_all.out"; $outfile="parsed/all_vs_all.parsed"; open (OUT, ">$outfile") || die "Cannot open file $outfile $!\n";
Обрабатываем результаты blast >more parsed/all_vs_all.parsed
Идентификация видов по номерам gi (gene identification) >perl extract_gi_numbers.pl >more gi_numbers.out Bordetella parapertussis 12822 | 3359.... 1616..... Desulfovibrio vulgaris DP4 | 1206..... 162139... Escherichia coli str. K-12 substr. MG1655 | 1612.... 4917.... 9011.... 2218..... 2265..... 1456..... 9454.... 3454..... 1577..... 3082..... 2885..... 1717..... 1613.... 6700.... 3334.... 2960..... 162135... Helicobacter pylori 26695 | 1564.... 1613..... 4873.... Rickettsia prowazekii str. Madrid E | 1560.... 1617.....
Отбираем суперсемейства, содержащие, по крайней мере, 4 вида. >perl parse_superfamilies_singlelink.pl 5 & Результат: > more parsed/all_vs_all.fam | wc -l 604 >perl simple_info.pl parsed/all_vs_all.fam Результат: parsed/all_vs_all.fam.info >perl sort_column.pl parsed/all_vs_all.fam.info Результат: parsed/all_vs_all.fam.info.sorted
Отбираем последовательности для суперсемейств >perl prepare_fa.pl parsed/all_vs_all.fam Результат: fa/fam_XX.fa Посмотреть содержимое: >less fa/fam_7.fa Проверить, что число найденных последовательностей совпадает с числом генов в суперсемействе: >more fa/fam_12.fa | grep '>' | wc -l 599
Выравнивание суперсемейств ~10 минут без больших суперсемейств Уберем большие суперсемейства в другую директорию >mkdir fa_big >mv fa/fam_12.fa fa_big/ > mv fa/fam_52.fa fa_big/ >mv fa/fam_98.fa fa_big/ >mv fa/fam_57.fa fa_big/ >mv fa/fam_58.fa fa_big/ >mv fa/fam_60.fa fa_big/ Запустим выравнивание >perl do_clustalw_aln.pl & Результат: dist/*.aln #! /usr/bin/perl -w #create 'dist' if it doesn't exist if (!opendir(DIR,"dist")){ mkdir("dist"); }else{ close(DIR); } while(defined($filename=glob("fa/*.fa"))) { print "$filename\n"; # clustalw each file system("clustalw -infile=$filename -align -type=protein"); } system("mv fa/*.aln dist/$d");
Построение деревьев методом расстояний с коррекцией Кимуры #! /usr/bin/perl -w # Tree reconstruction, using distance method with kimura correction # trees will be generated in the same directory 'dist' with extension *.ph while(defined($filename=glob("dist/*.aln"))) { # clustalw each file system("clustalw -infile=$filename -tree -OUTPUTTREE=dist -kimura"); } >do_clustalw_dist_kimura.pl Результат: dist/fam_##.ph Подготовка деревьев для BranchClust: >perl prepare_trees.pl Результат: trees/fam_##.tre
Обработка деревьев суперсемейств алгоритмом BranchClust > perl branchclust_all.pl 4 & Результаты: clusters/clusters_##.out clusters/family_##. Дополнительно: clusters_##.log >perl names_for_cluster_all.pl >perl detailed_summary.pl Результаты: detailed_summary.out families-names.list
tRNA synthases >more clusters/clusters_148.out.names
