Download

1 / 35
501 likes | 932 Views
Aldol and Claisen condensations. REACTION OF CARBON NUCLEOPHILES WITH CARBONYL GROUPS. some of the most useful synthetic methods for carbon-carbon bond formation. Robinson annulation. Wittig and related olefination reactions. REACTIONS OF CARBON NUCLEOPHILES WITH CARBONYL GROUPS.

E N D
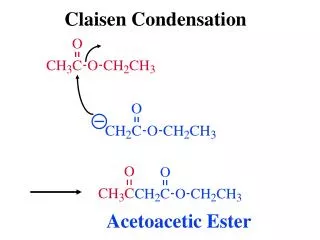
Aldol and Claisen condensations REACTION OF CARBON NUCLEOPHILES WITH CARBONYL GROUPS some of the most useful synthetic methods for carbon-carbon bond formation Robinson annulation Wittig and related olefination reactions REACTIONS OF CARBON NUCLEOPHILES WITH CARBONYL GROUPS The focus in these lessons is on some of the most useful synthetic methods for carbon-carbon bond formation : • the aldol and Claisen condensations • the Robinson annulation • the Wittig reaction and related olefination methods
the nature of X depends on the nature of A and B the product obtained the ways in which A, B and X interact All of these reactions begin by the addition of a carbon nucleophile to a carbonyl group.
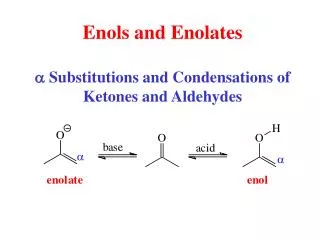
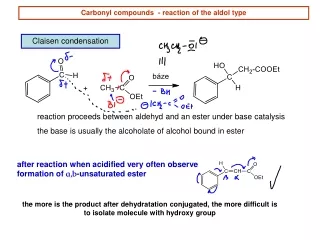
2.1. Aldol Addition and Condensation Reactions 2.1.1. The General Mechanism The prototypical aldol addition reaction is the acid- or base-catalyzed dimerization of a ketone or aldehyde. Under certain conditions, the reaction product may undergo dehydration leading to an a,b-unsaturated aldehyde or ketone. The mechanism of the base-catalyzed reaction involves equilibrium formation of the enolate ion, followed by addition of the enolate to a carbonyl group of the aldehyde or ketone.
In general, the reactions in the addition phase of both the base- and acid-catalyzed mechanisms are reversible. • The equilibrium constant for addition is usually unfavorable for acyclic ketones. • When the reaction conditions are sufficiently vigorous to cause dehydration, the overall reaction will go to completion, even if the equilibrium constant for the addition step is unfavorable. • The equilibrium constant for the dehydration phase is usually favorable, because of the conjugateda,b-unsaturated carbonyl system that is formed.
are aldol condensation involving two different carbonyl compounds are useful in synthesis whenthere is control on the carbonyl compound that acts as enolate precursor electrophile 2.1.2. Mixed Aldol Condensations with Aromatic Aldehydes mixed aldol reactions
One of the most general mixed aldol condensations involves the use of aromatic aldehydes with alkyl ketones or aldehydes. • Aromatic aldehydes are incapable of enolization and cannot function as the nucleophilic component. • Furthermore, dehydration is especially favorable because the resulting enone is conjugated with the aromatic ring. There are numerous examples of both acid- and base-catalyzed mixed aldol condensations involving aromatic aldehydes. The reaction is sometimes referred to as the Claisen- Schmidt condensation.
In the transition state for elimination to a cis double bond, an unfavorable steric interaction between the ketone substituent R and the phenyl group occurs. This interaction is absent in the transition state for elimination to the trans double bond. There is a pronounced preference for the formation of a trans double bond in the Claisen-Schmidt condensation of methyl ketones. This stereoselectivity arises in the dehydration step.
Additional insight into the factors affecting product structure was obtained by study of the condensation of 2-butanone with benzaldehyde. Under the reaction conditions used, it is not possible to isolate the intermediate ketols, because the addition step is rate-limiting. These intermediates have been prepared by alternative methods and put in acid or basic solution to observe their behaviour.
In basic solution • These results establish that the base-catalyzed dehydration is slow relative to the reverse of the addition phase for the branched-chain isomer. • In base, the straight-chain ketol is the only intermediate which is dehydrated. • The branched-chain ketol reverts to starting material.
> < In acid solution • Under acid conditions, both intermediates are dehydrated; however, the branched-chain ketol is formed most rapidly, because of the preference for acid-catalyzed enolization to give the more substituted enol.
In general, the product ratio of a mixed aldol condensation will depend upon the individual reaction rates. • Most ketones show a pattern similar to butanone in reactions with aromatic aldehydes. • Base catalysis favors reaction at a methyl position over a methylene group, whereas acid catalysis gives the opposite preference. ACID CATALYSIS BASE CATALYSIS
THE CHALLENGE THE CONTROL CHEMOSELECTIVITY Selectivity between different functional groups REGIOSELECTIVITY Control between different aspects of the same functional group STEREOSELECTIVITY Control over stereochemistry 2.1.3. Control of Regiochemistry and Stereochemistry of Mixed Aldol Reactions of Aliphatic Aldehydes and Ketones CONTROL = The confidence that molecules can be compelled to combine in the ways that we require
ENOLATE SILYL ENOL ETHER reacts with ELECTROPHILE IMINE ANION Lithium Enolates • Such reactions are normally carried out by complete conversion of the carbonyl compound that is to serve as the nucleophile to an enolate, silyl enol ether, or imine anion. The reactive nucleophile is then allowed to react with the second reaction component. CARBONYL COMPOUND = NUCLEOPHILE is converted in • As long as the addition step is faster than proton transfer, or other mechanisms of interconversion of the nucleophilic and electrophilic components, the adduct will have the desired structure. The term directed aldolreaction is given to these reactions.
In general, this requires that the product structure be controlled by kineticfactors, both in the formation of the enolate and in the addition step, and that equilibration by reversibility of either step be avoided. Such enolates are usually highly reactive toward aldehydes so that addition occurs rapidly when the aldehyde is added, even at low temperature. When the addition step is complete, the reaction is stopped by neutralization and the product is isolated. For example:
The guiding mechanistic concept for reactions carried out under these conditions is that they occur through a cyclic transition state in which lithium or another metal cation is coordinated to both the enolate oxygen and the carbonyl oxygen. This transition-state model has been the basis both for development of other reaction conditions and for the interpretation of the stereochemistry of the reaction.
Most enolates can exist as two stereoisomers. Also, most aldol condensation products formed from a ketone enolate and an aldehyde can have two diastereomeric structures. These are designated as syn and anti. The cyclic-transition-state model provides a basis for understanding the relationship between enolate geometry and the stereochemistry of the aldol product.
The enolate formed from 2,2-dimethyl-3-pentanone under kinetically controlled conditions is the Z-isomer. When it reacts with benzaldehyde, only the syn aldol is formed. This stereochemical relationship is explained by a cyclic transition state with a chair-like conformation. The product stereochemistry is correctly predicted if the aldehyde is in a conformation such that the phenyl substituent occupies an equatorial position in the cyclic transition state.
O O H O O H - + R O L i O L D A P h C H O + + R P h R P h - + R O L i R C H C H 3 3 R E Z anti syn ethyl 70 30 36 64 iso-propyl 40 60 18 82 tert-butyl 2 98 2 98 • A similar preference for formation of the syn aldol is found for other Z-enolates derived from ketones in which one of the carbonyl substituents is bulky. • Ketone enolates in which the other carbonyl substituent is less bulky show a decreasing stereoselectivity in the order • t-butyl > i-propyl > ethyl • This trend reflects a decreasing preference for formation of the Z-enolate.
E : Z Stereoselectivity LiTMP-LiBr LDA LiTMP 3.3 : 1 5 : 1 50 : 1 1.7 : 1 2 : 1 21 : 1 1 : >50 1 : >20 1 : >20 The E : Z ratio can be modified by the precise conditions for formation of the enolate. For example, the E : Zratio can be increased for 3-pentanone and 2-methyl-3-pentanone by use of a 1:1 LiTMP-LiBr mixture for kinetic enolization. The precise mechanism of this effect is not clear, but it probably is due to an aggregate species containing bromide acting as the base.
The enolates derived from cyclic ketones are necessarily E-isomers. The enolate of cyclohexanone reacts with benzaldehyde to give both possible stereoisomeric products under kinetically controlled conditions. The stereochemistry can be raised to about 6 : 1 in favor of the anti isomer under optimum condition.
Z-enolate syn aldol anti aldol E-enolate From these and related examples, the following generalizations have been drawn about kinetic stereoselection in aldol additions. • The chair transition-state model provides a basis for explaining the stereoselectivity observed in aldol reactions of ketones having one bulky substituent. The preference is • When the enolate has no bulky substituents, stereoselectivity is low. • Z-Enolates are more stereoselective than E-enolates. Because the aldol reaction is reversible, it is possible to adjust reaction conditions so that the two stereoisomeric aldol products equilibrate. This can be done in the case of lithium enolates by keeping the reaction mixture at room temperature until the product composition reaches equilibrium.
ANTI SYN This has been done, for example, for the product from the reaction of the enolate of ethyl t-butyl ketone and benzaldehyde. The greater stability of the anti isomer is attributed to the pseudoequatorial position of the methyl group in the chair-like chelate. With larger substituent groups, the thermodynamic preference for the anti isomer is still greater. Thermodynamic equilibration can be used to control product composition if one of the desired stereoisomers is significantly more stable than the other.
THE CONTROL OF ENOLATE STEREOCHEMISTRY THE ENHANCEMENT OF THE STEREOSELECTIVITY IN THE ADDITION STEP The requirement that an enolate have at least one bulky substituent restricts the types of compounds that can be expected to give highly stereoselective aldol additions. Furthermore, only the enolate formed by kinetic deprotonation is directly available. Ketones with one tertiary alkyl substituent give mainly the Z-enolate. However, less highly substituted ketones usually give mixtures of E- and Z-enolates. THE CHALLENGES
The enolates of other carbonyl compounds can be used in mixed aldol condensations. Extensive use has been made of the enolates of esters, thioesters, amides, nitriles, and nitroalkanes. • Because of their usefulness in aldol additions and other synthetic methods, there has been a good deal of interest in the factors that control the stereoselectivity of enolate formation from esters. • For simple esters such as ethyl propanoate, the E-enolate is preferred under kinetic conditions using a strong base such as LDA in THF solution. Inclusion of a strong cation solvating co-solvent, such as HMPA or tetrahydro-1,3-dimethyl-2(1H)pyrhidone (DMPU) favors the Z-enolate.
These observations are explained in terms of a cyclic transition state for the LDA/THF conditions and an open transition state in the presence of an aprotic dipolar solvent.
Simple alkyl esters show rather low stereoselectivity. However, highly hindered esters derived from 2,6-dimethylphenol or 2,6-di-t-butyl-4-methylphenol provide the anti stereoisomers.
The lithium enolates of a-alkoxy esters have been extensively explored, and several cases in which high stereoselectivity is observed have been documented. • This stereoselectivity can be explained in terms of a chelated ester enolate which is approached by the aldehyde in such a manner that the aldehyde R group avoids being between the a-alkoxy and the methyl group in the ester enolate. When the ester alkyl group R becomes very bulky, the stereoselectivity is reversed.
Boron Enolates Another important version of the aldol reaction involves the use of boron enolates. A cyclic transition state is believed to be involved, and, in general, the stereoselectivity is higher than for lithium enolates. The O-B bond distances are shorter than the O-Li bond in the lithium enolates, and this leads to a more compact transition state, which magnifies the steric interactions that control stereoselectivity.
Boron enolates can be prepared by reaction of the ketone with a dialkylboron trifluoromethanesulfonate (triflate) and a tertiary amine. The Z-stereoisomer is formed preferentially for ethyl ketones with various R‘ substituents. The resulting aldol products are predominantly the syn stereoisomers. The E-boron enolate from cyclohexanone shows a preference for the anti ketol product. The exact ratio of stereoisomeric ketols is a function of the substituents on boron and the solvent.
The E-boron enolates of some ketones can be preferentially obtained with the use of dialkylboron chlorides. Use of boron triflates with a more hindered amine favors the Z-enolate. The contrasting stereoselectivity of the boron triflates and chlorides has been discussed in terms of reactant conformation and the stereoelectronic requirement for perpendicular alignment of the hydrogen being removed with the carbonyl group.
Other methods are also available for generation of boron enolates. Diallylboranes react with acyclic enones to give Z-enolates by a 1,4-reduction. The preferred Z stereochemistry is attributed to a cyclic mechanism for hydride transfer: Z-Boron enolates can also be obtained from silyl enol ethers. This method is necessary for ketones such as ethyl t-butyl ketone, which gives E-boron enolates by other methods. The Z-stereoisomer is formed from either the Z- or E-silyl enol ether.
The E-boron enolates show a modest preference for formation of the anti aldol product. The general trend is that boron enolates parallel lithium enolates in their stereoselectivity but show enhanced stereoselectivity. They also have the advantage of providing access to both stereoisomeric enol derivatives.
Boron enolates can also be obtained from esters and amides and these too undergo aldol addition reactions. Various combinations of boronating reagents and amines have been used, and the E: Z ratios are dependent on the reagents and conditions. In most cases, esters give Z-enolates which lead to syn adducts, but there are exceptions.