Download

1 / 56
580 likes | 792 Views
Immunological bioinformatics. Ole Lund, Center for Biological Sequence Analysis (CBS) Denmark. World-wide Spread of SARS. Status as of July 11, 2003: 8437 Infected, 813 Dead. SARS. First severe infectious disease to emerge in the post-genomic era

E N D
Immunological bioinformatics Ole Lund, Center for Biological Sequence Analysis (CBS) Denmark.
World-wide Spread of SARS Status as of July 11, 2003: 8437 Infected, 813 Dead
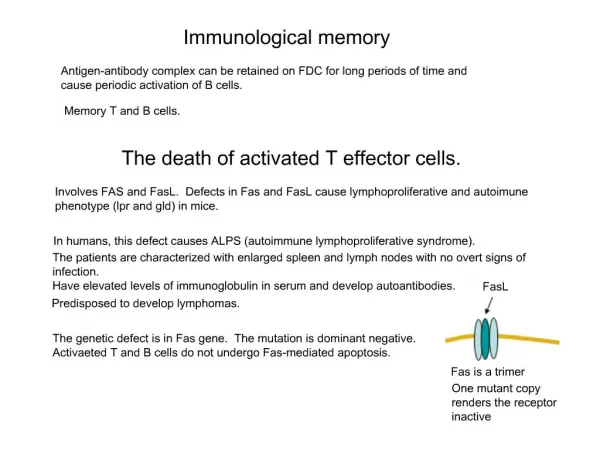
SARS • First severe infectious disease to emerge in the post-genomic era • Modern societies are vulnerable to epidemics • Classical containment strategies has been successful in controlling the epidemic, but • SARS may resurface (e.g. be seasonal) • Suggested existence of an animal reservoir could compromise the containment strategy • Need to develop a vaccine strategy • Biotechnology has provided new tools to analyze genome/proteome information and guide vaccine development. • The causative virus, the SARS corona virus (SARS CoV), has been isolated and full-length sequenced.
Main scientific achievements • Discovery of causative agent • Genome(s) • 3D Structure of main proteinase • Origin • Similar virus found in from Himalayan palm civets and other animals, including a raccoon-dog, and in humans working at an animal market in Guangdong, China (Guan et al., Sep 4, 2003). Himalayan (Masked) palm civet Ferret-Badger Raccoon-dog http://biobase.dk/~david-c/uk-dk-mammmal-list.htm
Source: Michael Buchmeier, Beijing June, 2003 New corona viruses 1978 Porcine Epidemic diarrhea virus (PEDV) Probably from humans 1984 Porcine Respiratory Coronavirus 1987 Porcine Reproductive and Respiratory Syndrome (PRRS) 1993 Bovine corona virus 2003 SARS
Will it be back? • When? • Every year?, Like the flu. • Every few years? Like measles used to. • Sporadic? Like Ebola • Never? • Lab safety: The patient, a 27-year-old virologist, worked on the West Nile virus in a biosafety level 3 lab at the Environmental Health Institute, where the SARS coronavirus was also studied (Enserink, 2003)
The immune system • The innate immune system • Found in animals and plants • Fast response • Complement, Toll like receptors • The adaptive Immune system • Found in vertebrates • Stronger response 2nd time • B lymphocytes • Produce antibodies (Abs) recognizes 3D shapes • Neutralize virus/bacteria outside cells • T lymphocytes • Cytotoxic T lymphocytes (CTLs) - MHC class I • Recognize foreign protein sequences in infected cells • Kill infected cells • Helper T lymphocytes (HTLs) - MHC class II • Recognize foreign protein sequences presented by immune cells • Activates cells
Weight matrices (Hidden Markov models) YMNGTMSQV GILGFVFTL ALWGFFPVV ILKEPVHGV ILGFVFTLT LLFGYPVYV GLSPTVWLS WLSLLVPFV FLPSDFFPS CVGGLLTMV FIAGNSAYE A2 Logo
Protein sequence information content • Entropy • Average Uncertainty in the random variable • H = -Spilog2pi range: 0 to log2(20) = 4.3 • Logo height I = log2(20) + H • Relative entropy (Kullback Leibler distance) • D = Spilog2(pi/qi) range: 0 to infinity • Mutual information • Reduction in uncertainty due to knowledge of another random variable (corresponds to correlation) • M = SSpijlog2(pij/pipj)
Prediction of MHC binding specificity • Simple Motifs • Allowed (non allowed) amino acids • Extended motifs • Amino acid preferences • Structural models • Limitations: precision of force field, and speed of calculations • Neural networks • Can take correlations into account
Log odds ratios • Used for scoring Alignments (BLAST), HMMs, Matrix methods • Odds ratio of observing given amino acids • Relative probability of observing amino acid i in motif position j • Oj = p(aai at pos j)/p(aai) • Assumption of independence => • Odds for observing sequence = O1O2 … On • Log odds ratio • LO = log(O1O2 … On) = log(O1)+log(O2)+…log(On) • LO in half bits = 2 LO/log(2)
G F C A
Evaluation of prediction accuracy Coverage = TP/actual_positive Reliability = TP/predicted_positive
The MHC gene region From Bill Paul, ”Fundamental Immunology”, 4th Ed
Human Leukocyte antigen (HLA=MHC in humans) polymorphism - alleles A total of 229 HLA-A 464 HLA-B 111 HLA-C class I alleles have been named, a total of 2 HLA-DRA, 364 HLA-DRB 22 HLA-DQA1, 48 HLA-DQB1 20 HLA-DPA1, 96 HLA-DPB1 class II sequences have also been assigned. As of October 2001 (http://www.anthonynolan.com/HIG/index.html)
HLA polymorphism - supertypes • Each HLA molecule within a supertype essentially binds the same peptides • Nine major HLA class I supertypes have been defined • HLA-A1, A2, A3, A24,B7, B27, B44, B58, B62 • Sette et al, Immunogenetics (1999) 50:201-212
HLA polymorphism - frequencies Supertypes Phenotype frequencies Caucasian Black Japanese Chinese Hispanic Average A2,A3, B27 83 % 86 % 88 % 88 % 86 % 86% +A1, A24, B44 100 % 98 % 100 % 100 % 99 % 99 % +B7, B58, B62 100 % 100 % 100 % 100 % 100 % 100 % Sette et al, Immunogenetics (1999) 50:201-212
Conclutions • We suggest to • split some of the alleles in the A1 supertype into a new A26 supertype • split some of the alleles in the B27 supertype into a new B39 supertype. • the B8 alleles may define their own supertype • The specificities of the class II molecules can be clustered into nine classes, which only partly correspond to the serological classification Lund O, Nielsen M, Kesmir C, Petersen AG, Lundegaard C, Worning P, Sylvester-Hvid C, Lamberth K, Roder G, Justesen S, Buus S, Brunak S. Definition of supertypes for HLA molecules using clustering of specificity matrices. Immunogenetics. 2004 Feb 13 [Epub ahead of print]
MHC class I binding of SARS peptides • Predictions for all supertypes • Broad population coverage • Allele specific neural networks • Peptides with associated measured binding affinity • A1 (A0101), A2 (A0204), A3 (A1101+A0301), B7 (B0702) • Weight matrices • Peptides from public databases (Sypfeithi, MHCpep) • A24, B27, B44, B58 and B62
Super type weight matrices B27 B44 B58 B62
Epitope predictions • Binding to MHC class I • High probability for C-terminal proteasomal cleavage • No sequence variation
Inside out: • Position in RNA • Translated regions (blue) • Observed variable spots • Predicted proteasomal cleavage • Predicted A1 epitopes • Predicted A*0204 epitopes • Predicted A*1101 epitopes • Predicted A24 epitopes • Predicted B7 epitopes • Predicted B27 epitopes • Predicted B44 epitopes • Predicted B58 epitopes • Predicted B62 epitopes
Peptide-MHC complex Incubation Development Strategy for the quantitative ELISA assayC. Sylvester-Hvid, et al., Tissue antigens, 2002: 59:251 • Step I: Folding of MHC class I molecules in solution b2m Heavy chain peptide • Step II: Detection of de novo folded MHC class I molecules by ELISA
Summery of peptide binding assays #tested #binding <500nM A1 15 13 A2 15 12 A3 15 14 A24 0 - B7 15 10 B27 13 2 B44 0 - B58 15 13 B62 14 12
Initial polytope (19 HIV epitopes) • New epitopes 12 • Poor C-term cleavage 8 • Cleavage within 31 • Linker length 12
Optimized polytope • New epitopes 1 • Weak C-term cleavage 3 • Cleavage within 7 • Linker length 37
Virtual matrices • HLA-DR molecules sharing the same pocket amino acid pattern, are asumed to have identical amino acid binding preferences.
MHC Class II binding • Virtual matrices • TEPITOPE: Hammer, J., Current Opinion in Immunology 7, 263-269, 1995, • PROPRED: Singh H, Raghava GPBioinformatics 2001 Dec;17(12):1236-7 • Web interface http://www.imtech.res.in/raghava/propred • Prediction Results
Complexity of problem Peptides of different length Weak motif signal Alignment crucial Gibbs Monte Carlo sampler RFFGGDRGAPKRG YLDPLIRGLLARPAKLQV KPGQPPRLLIYDASNRATGIPA GSLFVYNITTNKYKAFLDKQ SALLSSDITASVNCAK PKYVHQNTLKLAT GFKGEQGPKGEP DVFKELKVHHANENI SRYWAIRTRSGGI TYSTNEIDLQLSQEDGQTIE MHC class II prediction
Class II binding motif Random ClustalW Alignment by Gibbs sampler RFFGGDRGAPKRG YLDPLIRGLLARPAKLQV KPGQPPRLLIYDASNRATGIPA GSLFVYNITTNKYKAFLDKQ SALLSSDITASVNCAK PKYVHQNTLKLAT GFKGEQGPKGEP DVFKELKVHHANENI SRYWAIRTRSGGI TYSTNEIDLQLSQEDGQTI Gibbs sampler
Polytope construction Linker NH2 M COOH Epitope cleavage C-terminal cleavage New epitopes Cleavage within epitopes