Download

1 / 19
190 likes | 279 Views
EDACC Primary Analysis Pipelines. Cristian Coarfa Bioinformatics Research Laboratory Molecular and Human Genetics. Data Levels. ChIP-Seq Shotgun Bisulfite Sequencing Methyl-C Reduced Representation Bisulfite Sequencing RRBS MRE-Seq MeDIP-Seq Chromatin Accessibility

E N D
EDACCPrimary Analysis Pipelines Cristian Coarfa Bioinformatics Research Laboratory Molecular and Human Genetics
ChIP-Seq Shotgun Bisulfite Sequencing Methyl-C Reduced Representation Bisulfite Sequencing RRBS MRE-Seq MeDIP-Seq Chromatin Accessibility small RNA-Seq mRNA-Seq Data Types Submitted To EDACC
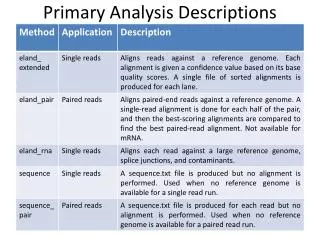
Common processing step to all pipelines High throughput Sequence space: Illumina Color space: SOLID Quick and accurate anchoring Reads size varies 36-76 bp Short read aligners 1st generation: Maq, soap Ungapped alignment 2nd generation: bowtie, bwa, soap 2 Tradeoff speed for sensitivity, good enough for many applications Mapping tools Robust to indels Sensitive to variable number of mismatches Read Mapping
Positional Hashing Regular reads mapping Bisulfite sequencing mapping Integrate basepair variation with epigenetic variation SAM output, easy integration with other analysis tools Accuracy without sacrificing efficiency Pash 3.0
Current tools: BSMAP, RMAP-BS, mrsFast, Zoom Pash 3.0 Integrate mutation discovery with basepair-level methylation discovery Speedup General approach Covert C’s to T’s in reads and/or reference Use mappings, reads and reference to determine methylated sites Pash 3 Generate and hash all possible kmers for reads CTT: CCC, CCT, CTC, CTT Map against forward and reverse complement chromosome strands Superior sensitivity to other tools, without loss of efficiency Bisulfite Sequencing
Developed at Penn State University Benefits Rapid deployment tool Share pipelines w/ others Alan Harris, Sriram Raghuram Deployed Galaxy/Genboree Integration w/ Genboree API for upload/download Adaptors for LFF file format support EDACC XML validation tools Sriram Raghuram, Andrew Jackson, Cristian Coarfa Integration with compute clusters Arpit Tandon, Sriram Raghuram Deployed analysis tools Galaxy/Genboree http://genboree.org/galaxy
Implemented & exposed via Galaxy/Genboree Read mapping Bisulfite Sequencing read mapping Peak calling (ChIP-Seq, MeDIP-Seq) MACS (Harvard), FindPeaks (UBC) Chromatin accessibility HotSpot (UW) Small RNA-seq Coming soon mRNA seq Expression, alternative splicing Gene fusion Typical user interaction Use Galaxy for user input Submit jobs to a cluster Upload results to Genboree Primary Analysis Pipelines
Select uniquely mapping reads Build read density maps Extend each read 200bp along the mapping strand Remove monoclonal reads Generate WIG data Can be visualized in Genboree and UCSC Peak calling FindPeaks, MACS Intepret Peaks Overlap with genomic features of interest: gene promoters, etc ChIP-Seq
Select uniquely mapping reads Build read density maps Determine methylated CpGs FindPeaks MeDIP-Seq
Select uniquely mapping reads Determine unmethylated CpGs MRE-Seq
Shotgun Bisulfite Sequencing Methyl-C Genome wide Reduced Representation Bisulfite Sequencing RRBS Enzyme cocktail Map using Pash Build methylation maps Bisulfite Sequencing
Methylation Maps Position Strand CHHStatus Methylation Unmethylated TotalReads 50100242 + CG 1 0 1 50100243 - CG 40 11 51 50100250 + CG 1 0 1 50100251 - CG 37 8 46
Trim adapters Map reads onto target genome up to 100 locations per read Interpret Overlap w/ miRNAs, piRNAs, sno/scaRNAs Small RNA-Seq
Download the input MeDIP-Seq file from the workshop wiki Analyze it using FindPeaks in Galaxy Obtain results in Genboree Lff format Upload the results to Genboree database View the results in a tabular view Find the largest peaks Explore them in the Genboree browser Exercise