Download
1 / 22
220 likes | 235 Views
Explore 4-hop classification using IRIS attributes - Sepal Length, Sepal Width, Petal Length, Petal Width. Try various rules and algorithms to classify IRIS dataset confidently. Discover decision trees for accurate classification. Analyze patterns for classifying Iris flowers.

E N D
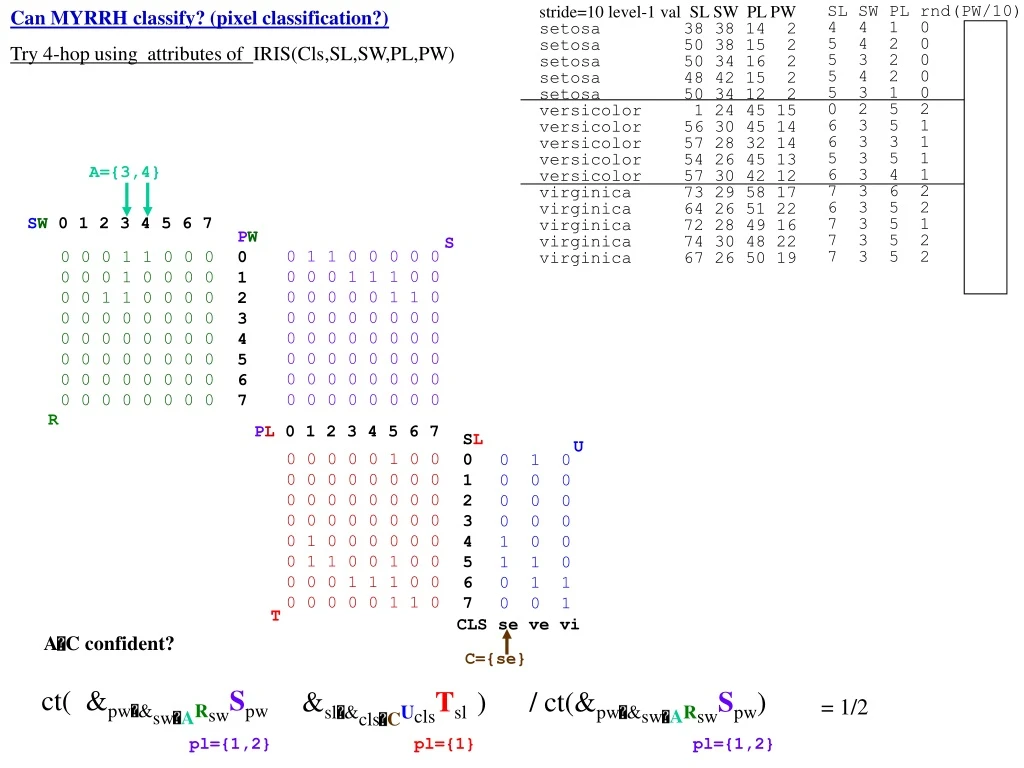
SL SW PL rnd(PW/10) 4 4 1 0 5 4 2 0 5 3 2 0 5 4 2 0 5 3 1 0 0 2 5 2 6 3 5 1 6 3 3 1 5 3 5 1 6 3 4 1 7 3 6 2 6 3 5 2 7 3 5 1 7 3 5 2 7 3 5 2 Can MYRRH classify? (pixel classification?) Try 4-hop using attributes of IRIS(Cls,SL,SW,PL,PW) A={3,4} stride=10 level-1 val SL SW PL PW setosa 38 38 14 2 setosa 50 38 15 2 setosa 50 34 16 2 setosa 48 42 15 2 setosa 50 34 12 2 versicolor 1 24 45 15 versicolor 56 30 45 14 versicolor 57 28 32 14 versicolor 54 26 45 13 versicolor 57 30 42 12 virginica 73 29 58 17 virginica 64 26 51 22 virginica 72 28 49 16 virginica 74 30 48 22 virginica 67 26 50 19 C={se} ct( &pw&swARswSpw &sl&clsCUclsTsl ) / ct(&pw&swARswSpw) pl={1,2} pl={1} pl={1,2} SW 0 1 2 3 4 5 6 7 PW 0 1 2 3 4 5 6 7 S 0 1 1 0 0 0 0 0 0 0 0 1 1 1 0 0 0 0 0 0 0 1 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 1 0 0 0 0 0 0 1 0 0 0 0 0 0 1 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 R PL 0 1 2 3 4 5 6 7 SL 0 1 2 3 4 5 6 7 U 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 1 1 0 0 1 0 0 0 0 0 1 1 1 0 0 0 0 0 0 0 1 1 0 0 1 0 0 0 0 0 0 0 0 0 0 1 0 0 1 1 0 0 1 1 0 0 1 T CLS se ve vi AC confident? = 1/2
SL SW PL rnd(PW/10) 4 4 1 0 5 4 2 0 5 3 2 0 5 4 2 0 5 3 1 0 0 2 5 2 6 3 5 1 6 3 3 1 5 3 5 1 6 3 4 1 7 3 6 2 6 3 5 2 7 3 5 1 7 3 5 2 7 3 5 2 A={3,4} 1-hop: IRIS(Cls,SL,SW,PL,PW) stride=10 level-1 val SL SW PL PW setosa 38 38 14 2 setosa 50 38 15 2 setosa 50 34 16 2 setosa 48 42 15 2 setosa 50 34 12 2 versicolor 1 24 45 15 versicolor 56 30 45 14 versicolor 57 28 32 14 versicolor 54 26 45 13 versicolor 57 30 42 12 virginica 73 29 58 17 virginica 64 26 51 22 virginica 72 28 49 16 virginica 74 30 48 22 virginica 67 26 50 19 C={se} PW 0 1 2 3 4 5 6 7 PL 0 1 2 3 4 5 6 7 SL 0 1 2 3 4 5 6 7 CLS se ve vi CLS se ve vi CLS se ve vi SW 0 1 2 3 4 5 6 7 CLS se ve vi 0 2 3 0 0 0 0 0 0 0 0 1 1 3 0 0 0 0 0 0 0 4 1 0 0 0 0 0 1 4 0 0 1 0 0 0 0 1 3 0 0 0 0 0 0 0 1 4 5 0 0 0 0 0 0 0 0 4 1 0 0 0 0 0 0 1 4 0 0 0 0 0 0 0 0 2 3 0 0 0 0 0 1 4 0 0 0 0 0 0 0 5 0 0 0 0 / \ PW=0 else | se PL{3,4} & SW=2 & SL=5 else | ve 2 of 3 of: else PL{3,4,5} | SW={2,3} vi SL={5,6} | ve SW 0 1 2 3 4 5 6 7 CLS se ve vi 0 0 0 1 1 0 0 0 0 0 1 1 0 0 0 0 0 0 0 1 0 0 0 0 R 1-hop AC is more confident: ct(RA&cls{se}Rcls) / ct(RA) = 1 sw= {3,4} sw= {3,4} sw= {3,4} But what about just taking R{class}? Gives {3,4}se {2,3}ve {3}vi This is not very differentiating of class. Include the other three? {4,5}se {5,6}ve {6,7}vi These rules were derived from the binary relationships only. A minimal Decision Tree Classifier suggested by the rules: {3,4}se {2,3}ve {3}vi {1,2}se {3,4,5}ve {5,6}vi {0}se {1,2}ve {1,2}vi I was hoping for a "Look at that!" but it didn't happen ;-)
SL SW PL rnd(PW/10) 4 4 1 0 5 4 2 0 5 3 2 0 5 4 2 0 5 3 1 0 0 2 5 2 6 3 5 1 6 3 3 1 5 3 5 1 6 3 4 1 7 3 6 2 6 3 5 2 7 3 5 1 7 3 5 2 7 3 5 2 A={1,2} 2-hop stride=10 level-1 val SL SW PL PW setosa 38 38 14 2 setosa 50 38 15 2 setosa 50 34 16 2 setosa 48 42 15 2 setosa 50 34 12 2 versicolor 1 24 45 15 versicolor 56 30 45 14 versicolor 57 28 32 14 versicolor 54 26 45 13 versicolor 57 30 42 12 virginica 73 29 58 17 virginica 64 26 51 22 virginica 72 28 49 16 virginica 74 30 48 22 virginica 67 26 50 19 sl={4,5} sl={4,5} sl={4,5} C={se} PL 0 1 2 3 4 5 6 7 SL 0 1 2 3 4 5 6 7 U 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 1 3 0 0 1 0 0 0 0 0 1 1 2 0 0 0 0 0 0 0 3 1 0 0 1 0 0 0 0 0 0 0 0 0 0 1 0 0 4 1 0 0 3 1 0 0 4 T CLS se ve vi ct(ORplATpl &clsCUcls) / ct(ORplATpl) =1 Mine out all confident se-rules with minsup = 3/4: Closure: If A{se} is nonconfident and AUse then B{se} is nonconfident for all B A. So starting with singleton A's: ct(Tpl=1 &Use) / ct(Tpl=1) = 2/2 yes. A= {1,3} {1,4} {1,5} or {1,6} will yield nonconfidence and AUse so all supersets will yield nonconfidence. ct(Tpl=2 &Use) / ct(Tpl=2) = 1/1 yes. A= {1,2} will yield confidence. ct(Tpl=3 &Use) / ct(Tpl=3) = 0/1 no. A= {2,3} {2,4} {2,5} or {2,6} will yield nonconfidence but the closure property does not apply. ct(Tpl=4 &Use) / ct(Tpl=4) = 0/1 no. ct(Tpl=5 &Use) / ct(Tpl=5) = 1/2 no. ct(Tpl=6 &Use) / ct(Tpl=6) = 0/1 no. etc. I conclude that this closure property is just too weak to be useful. And also it appears from this example that trying to use myrrh to do classification (at least in this way) does not appear to be productive.
Back to the classical collaborative filtering problem (AKA: customer preference predictor) which is critical for on-line retailing (e.g., Netflix Cinematch, Amazon's suggester, Yahoo's, etc.). On the face of it, it is a classical classification (based on purchase or rating history what rating would customer, c, give to Item, i? - that is, "In what class [rating] would c put i?". Let's assume a 5 star rating system. (There is always 2 stars - buy/didn't_buy) 2 2 2 3 3 3 4 4 4 5 5 5 I I C C 2(I,C) 4(I,C) 0 0 0 0 0 0 0 1 4 4 0 0 1 0 0 0 0 0 3 3 1 0 0 0 0 1 0 1 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 1 0 0 0 0 1 0 0 2 2 0 0 0 1 1 0 0 0 1 1 3(I,C) 5(I,C) I 0 1 0 0 4 3 0 0 1 1 2 0 0 0 0 1 0 0 0 1 C 1(C,I) Can MRRYH help? To predict Rating(c,i):
Tools are needed that can identify binding sites on proteins, which consist of residues at the interface of the interactions. The plan is to develop computational methods that can automatically predict binding sites on the protein structure and that can help to gain insights on the mechanistic basis of the interactions. The focus is on binding sites corresponding to protein-protein interactions and protein-nucleic acid interactions. We will refer to these binding sites as macromolecule-binding sites, since both proteins and nucleic acids are macromolecules. In practice, many researchers predict binding sites by searching for occurrences of conserved sequence or structural motifs or transferring binding site information from homologous proteins using sequence or structural alignment. Altho this has been success in many applications, they cannot apply to orphan proteins, which have no homologs available. Furthermore, the development of biologically significant structural motifs usually requires extensive interference from experts and is hard to automate. Consequently, there are only a few structural motifs available today. The guiding hypothesis for this projectis that stable interactions between macromolecules require the binding sites to possess favorable binding conditions in multiple aspects, including geometric complementarities, evolutionary conservation, hydrophobic force, electrostatic force and other physical and chemical forces. A solution must assess multiple features covering different aspects of the protein surface in order to predict binding sites. The overal goal of this project is to develop data models and computational methods for binding site prediction and analysis. Specific Aim 1: Develop expressive graph models for proteins. Proteins fold into a three-dimensional (3D) structure in cells, which is pivotal for proteins to perform their function. Graph models will be developed for representing crucial structural information and the spatial distribution of multiple features on the protein structure. Residues will be represented as graph vertices. Sequence and structural motifs, evolutionary profiles, physical and chemical properties will be encoded w vertex labels The spatial arrangement of residue side chains will be represented using three angles and one distance value that uniquely define the spatial relationship between two side chains. These angles and distance value will be shown as edge labels on the graphs. The flexibility of the protein structure will also be encoded into the graphs. Specific Aim 2: Develop new graph kernel methods for binding site prediction. Graph comparison is computationally intensive. In the proposed graph models, vertices and edges are labeled with real numbers, which adds difficulty. We will use a new type of graph kernel to exploit the rich info encoded in the graph models for binding site prediction. The new graph kernels resolve the tottering problem and other weaknesses associated with current graph kernels. The graph kernels will be embedded in machine-learning methods to learn complex relationships for binding site predictions. Specific Aim 3: Knowledge discovery in binding sites. Evidence shows macromolecule-binding sites have a modular org. Kernel clustering will be used to discover modules in the binding sites and investigate how modules evolve in the evolution and how they interact with each other. Kernel principle component analysis to identify characteristic patterns associated with different modules and different categories of binding sites. These patterns can be viewed as motifs. However, they differ from traditional sequence/structural motifs in that they integral multiple features and they don't require alignment of homologous proteins.
To predict binding sites on a protein is to identify the residues at the interface of the interactions. In practice, many researchers look for binding sites by searching for occurrences of sequence or structural motifs or by transferring binding sites information from homologous proteins using sequence or structural alignment. Altho these have achieved success in many apps, they cannot apply to orphan proteins, which have no homologs available. Constructing structural motifs relies on structural multiple alignment, needing manual interference and is hard to automate. In the past, researchers have tried to develop automatic methods using machine-learning for binding site prediction. However, only limited success has been achieved. The first challenge is in the data representation of the protein structure. The protein structure is a 3-dimensional (3D) object, which no current machine-learning methods can directly deal with. Thus, the 3D structures must be reduced to 2D objects, e.g. graphs, or 1D objects, e.g. vectors. The challenge here is that the reduction operation must not discard structural information necessary for the interactions. In other words, the 2D (or 1D) rep. models much contain sufficient structural information to enable binding site prediction. The graph models are designed to maintain structural info for binding site prediction, incl. spatial arrangement of residue side chains, local structure environment of residues, and spatial distribution of multiple physical chemical properties. The model represents the spatial arrangement of a pair of residue side chains using three angles and a distance. Another obstacle in the data representation of proteins is the flexibility of protein structures. Proteins may undergo confirmation change in binding so structure flexibility must be considered in predicting binding sites. However, due to technical difficulties, no previous graph model has addressed the protein flexibility problem. The proposed work is also innovative in that the proposed graph models take into account protein flexibility. The side-chain arrangement, local structural environment, and contacting relationship between residues are defined depending on the protein flexibility. The second challenge for automatic binding site prediction lies in the machine-learning methods. he machine-learning methods must have the ability to fully exploit the information in the data models to discover complex relationships. In the proposed work, the data are graphs labeled with multiple continuous features. Current machine-learning methods are not sufficient to handle this type of data. The innovation of the proposed work also resides in the new graph kernels that we propose to develop. The new graph kernels use innovative ideas to solve the tottering problem and other weaknesses of current methods. The proposed work is innovative because we use an innovative approach to investigate the modular organization of binding sites and discover characteristic patterns associated with modules and different types of binding sites. Currently no effective methods are available for tackling these problems. The graph models and graph kernel methods develop in this work make it feasible to perform the analyses.
BACKGROUND AND SIGNIFICANCE Persistent pathogens pose a great threat to public health. We will develop and use bioinformatics tools to help understand the structural and mechanistic basis of pathogen persistence. In molecular biology, there is an urgent need for tools that can identify binding sites on proteins, which consist of residues at the interface of the interactions. These residues contribute to the affinity of the interactions and provide target for drug design. We propose to develop novel computational methods that can automatically predict binding sites on the protein structure and that can help gain insights on the mechanistic basis of the interactions. We will use these novel methods to help elucidate the function of proteins. We will focus on the binding sites that correspond to protein-protein interactions and protein-nucleic acid interactions. We will refer to this type of binding sites as macromolecule-binding sites, since both proteins and nucleic acids are macromolecules. Unlike ligand-binding sites that often occur in pockets on the protein surface, macromolecule-binding sites usually locate at large planar surfaces. Thus, although numerous pocket-based methods have been developed for ligand-binding site identification, the prediction of macromolecule-binding sites cannot take the same approach. The binding site prediction problem is also different from hot-spot prediction, in which only residues with high contribution to the binding affinity are of interest, or catalytic site residues prediction, in which only residues that directly perform the catalytic activity are predicted. Many methods have been developed for macromolecule-binding site prediction. At one level, they can be divided into two groups: those rely on structural templates and those use automatic methods to analyze various features on the binding sites to discover predicting patterns. The first group of methods derives consensus structural templates of binding sites from a set of protein structures that have the same function. Then, new protein structures are scanned to search for the occurrences of the templates1-9. In some studies, structural templates are used in combination with other features such as electrostatic potentials10 and sequence profiles11,12. This group cannot apply to orphan proteins with no homologs available. The main drawback is the difficulty in deriving structural templates. Some derive structural templates by multiple alignment of protein structures6,10. This approach is hard to automate to produce objective results. Altho common patterns can be generated automatically from protein structure alignment at fold and topology levels13,14, generating structural templates reflecting local structural similarities still requires manual adjustments i.e., setting the anchor point of the multiple alignment. To date, there is no automated objective method for deriving structural templates of binding sites using multiple alignment of protein structures. Others identified structural templates by detecting recurring subgraphs15-18. However, these methods can only be used to find small templates, typically less than 6 residues, due to demanding computation requirement in generating all possible subgraphs in all protein structures. These methods usually report too many patterns. Enormous efforts by experts are needed to identify biologically significant patterns. The second group of methods uses machine-learning to analyze various features on binding sites to discover predicting patterns19-35. A wide range of features, including amino acid identity, sequence profile, evolutionary conservation, solvent accessibility, structural curvature, pocket size, electrostatic potentials and predicted secondary structure, have been analyzed. In these studies, the features of a surface patch are represented as a vector of values and inputted to a machine-learning method. This group of methods has the advantages that: (1) they can automatically discover sophisticated attribute-function relationships; (2) they also consider many features other than geometry that are important for the interactions; and (3) they can capture weak relationships that do not necessarily exist in every protein. However, as of today, this group of methods still suffer low accuracy36. The low accuracy is mainly due tothe use of vectors to represent surface patches. When the features of a surface patch are encoded using a vector, the information of how these features distribute on the structure is lost.
Introduction In this project, the Co-PI will develop graph models for protein representation. The graph models will maintain crucial structural information and encode the spatial distribution of multiple features on the proteins. Then, the Co-PI will develop new graph kernel methods that can fully exploit the rich information contained in the graphs for binding site prediction. Justification and Feasibility Stable interactions between macromolecules require the binding sites to possess favorable conditions in multiple aspects, including geometric complementarities, evolutionary conservation, hydrophobic force, electrostatic force and other physical and chemical forces. Thus, a method must assess features covering these aspects of the proteins in order to identify the binding sites. The graph models proposed in this work incorporate a wide range of features covering these aspects, and the proposed graph kernels combined with machine-learning methods are capable of discover complex patterns in the proposed graph models. Thus, we believe that the proposed methods can achieve success in binding site prediction and analysis. To test the feasibility of the proposed approach, we conducted a preliminary study on using graph kernel method to predict DNA-binding sites. We used a dataset of 171 DNA-binding proteins collected in our previous study19. We divided the protein surface into overlapping patches, such that each patch included a surface residue and its neighboring residues. Each patch was assigned to either positive class or negative class depending on whether the center residue was in the binding sites. Then, each patch was represented as a graph, such that each amino acid residue was represented using a vertex and an edge was added between two vertices if the corresponding residues were within a distance of 3.5 Å. Each vertex was then labeled with six features of the corresponding amino acid residue, including residue identity, sequence conservation score, structural conservation score, solvent accessibility, electrostatic potential and surface curvature. We used a shortest-path graph kernel to calculate the similarity between graphs. For more details about the shortest-path graph kernels, please see section C.3.ii.d. Briefly, a shortest-path graph kernel method compares all-pairs shortest paths between graphs. The comparison of two paths includes the comparison of path length and source and destination vertices. A Gaussian function was used to compare vertices based on the vertex labels. A Brownian kernel was used to compare the path length. The graph kernel was embedded into a support vector machine (SVM) to build a predictor for DNA-binding site prediction. When evaluated using leave-one-out cross-validation, the predictor achieved 89% accuracy, 90% specificity and 88% sensitivity. We also evaluated how each of the six features affected the prediction performance. When the feature number increased from one to six, the accuracy gradually increased from 86% to 89%. To further evaluate the method, we tested it using an independent set of 13 proteins used in a previous study21 whose apo-state structure (i.e. unbounded with DNA) and holo-state structure (i.e. bounded with DNA) were available. We used the predictor to predict DNA-binding sites on the apo-state structures and the predictions were compared against the actual binding sites gleaned from the holo-state structures. For each test protein, we ranked the surface patches by the prediction score from high to low. Remarkably, the top 1 patch in all of the 13 proteins belonged to the actual DNA-binding sites. This preliminary study shows that the graph kernel approach is able to discover predictive patterns on DNA-binding sites. The results are very encouraging. The top 1 prediction patch accurately indicates the location of the DNA-binding sites in all independent test proteins. This level of success would allow the method to make significant contribution in real applications.
Specific Aim 1: Develop expressive graph models for proteins In the basic model, each amino acid residue is represented using a vertex and an edge is added between two vertices if the corresponding residues are within a certain distance. Each vertex is labeled with a set of features associated with the corresp. amino acid residue. Each edge is labeled with an edge label. We will explore the following techniques to refine the graph representation. Arrangement of residue side chains. For any residue pair, the relative arrangement of their side chains in the3D space can be defined using three angles and a distance as illustrated in Figure 1. For each residue, we draw a line to connect the Caatom and the mass center (MC) of the side chain. Let's refer to this line as side_chain line. Then, we draw another line to connect the Ca atoms of the two residues. Let's refer to this line as center line. a and b are the angles between the center line and the side_chain line for residue i and j respectively (Figure 1A). Let's rotate the molecule so that the center line is perpendicular to the paper as shown in Figure 1B. g is the angel defined by the side_chain lines of the two residues. Let d be the distance between the two Caatoms. The vector <a, b, g, d> will uniquely define the relative arrangement of the 2 side chains. For each pair of contacting residues, we will use <a, b, g, d> to label the edge between them. Local structural environment of a residue influences how the residue functions. We will represent it using a vector of 20 values, corresp. to the counts of 20 types of residues within a sphere centered at the residue. Or, we can count functional groups, or atom types within the sphere. The radius of the sphere is a parameter to be decided. The vector will be used to label the vertex corresponding to the residue. Conserved motifs. Numerous sequence and structural motifs have been created by experts and made available on public databases7-9,37. These motifs can provide insightful information about binding sites. For a given protein, we will search for occurrences of motifs using the searching engines available at the corresponding motif databases. If a motif is found, then all the residues involved in the motif will be labeled with the ID of the motif. A residue could be labeled with multiple motif IDs. Evolutionary profile. Evolutionary properties have been widely used to predict binding sites38-40. For each protein, we will use BLAST41 to search for similar sequences in the NCBI nr database to generate a position specific scoring matrix (PSSM). Each residue is then labeled with a row of values from the PSSM that correspond to the residue. Conservation score will also be calculated based on the alignment. Reduced alphabets. There are 20 types of amino acids, some share similar physicochemical properties. Many cluster amino acids into groups based on different criteria and developed reduced alphabets for representating amino acids42-45. Using reduced alphabets, researchers have achieved improved performance in function and structure predictions42,46,47. We will explore various reduced alphabets to represent the identity of residues. Each vertex will be given a label showing the identity of the corresponding residue. Figure 1. Relative arrangement of two side chains is uniquely defined by three angels and one distance. Ca-i: Ca atom of residue i. MC-i: Mass center (MC) of side chain i. Ca-j: Ca atom of residue j. MC-j: Mass center (MC) of side chain j. A. a and b denote the angles between the center line and the side_chain line for residue i and j respectively. d is the distance between the Caatoms. B. The molecule is rotated so that the center line is perpendicular to the paper (Ca-i overlaps with Ca-j). g is the angel defined by the two side_chain lines. Graph edges. We will explore 3 types of edges used in the analysis of the protein structure, including distance-based edges, in which an edge is added if the distance between two amino acids is less than a threshold, Delaunay edges from the Delaunay tessellation, and almost-Delaunay edges, which are an extension of Delaunay edges by taking into account the perturbation or motion of point coordinates48. Geometrical, physical, and chemical properties. We systematically explore a wide range of features to select a set that are crucial for binding site prediction.The features to be explored include but are not limited to solvent accessibility, structure curvature, pocket size, b-factor, packing density, electrostatic potentials and the many features associated with the amino acids as indexed in the AAindex49.
Expected outcomes of Specific Aim 1: an expressive rep. model for proteins. The model is expressive in the sense that it encodes rich info about proteins. The model efficiently represents the relative arrangement between pairs of amino acid side chains using 3 angles and 1 dist. The side chain has the functional groups of amino acids. A proper arrangement of side chains in the binding site is required for stable interactions. By encoding the relative arrangement between every side chain pair, it captures the overall arrangement of all side chains in the binding sites. A challenge in representation for protein structures is to maintain useful structure info when reducing the 3D structure to a 2D or 1D object. As far as we know, no previous graph model has been able to represent the spatial arrangement of side chains. Additionally, the proposed model also incorporate conserved motifs. Although, motifs have been used widely to predict protein function and functional site, graph models used in previous studies have not tried to use this crucial information. In summary, the proposed model incorporates multiple features that describe different aspects of the protein, including sequence, structure, evolutionary properties, and chemical and physical properties. With such rich information, the proposed model will provide a solid foundation for using machine-learning methods to predict and analyze binding sites in Specific Aims 2 and 3. Anticipated problems and alternatives of Specific Aim 1 Flexibility of protein structure. Proteins may undergo confirmation change upon binding. However, the proposed models are rigid. Recent reports have shown that residues at protein-protein interfaces have lower mobility than other residues on the protein surface50,51. Thus, we expect proposed model to be successful in modeling most proteins. In case it fails, we will explore alternatives that explicitly address the protein flexibility problem. For a given protein structure, we will first do a dynamics simulation using NAMD52 or Gromacs53 to get n snapshots of the structure, where n is a parameter to be explored. Then, the mobility of a residue can be calculated by superimposing the snapshots to calculate the RMSD for it. In the graph model, we will assign edges between residues using a distance threshold that depends on the mobility of residues. E.g., we can use c+a(d1+d2), where c is a constant, a is parameter, d1 and d2 represent the mobility of the involving residues. Using this, two residues separated by a distance greater than the constant c still have chance to form a contact if they have high mobility. Or, determine whether 2 residues contact in each of the n snapshots using a constant threshold and then calculate the frequency of their contacts. If the frequency is high, we can assign an edge between them and label the edge with the frequency. We will also take into account protein flexibility in the calculation of local structural environment and side-chain arrangement. The local structural environment of a residue will take the average local structural environment of it in the n snapshots. The values of a, b, g and d in the side-chain arrangement will each be replaced by two values that denote the minimum and maximum value observed in the snapshots. Molecular dynamics simulation could be very time-consuming. To address this problem, we can use snapshots of protein structure from the protein confirmation data deposited in many protein dynamics databases, like Database of Macromolecular Movements54,Dynameomics55, and DynDom56. Feature selection and dimension reduction.In the proposed model, we have put a large number of features on the edge labels and vertex labels. While a large number of features can encode rich information for the analysis, it may also require long running time in the following prediction and analysis. Furthermore, some features maybe redundant. Thus, we will refine the model by reducing the dim of the features needed for the graph labels. We can filter out redundant features by performing Pearson's correlation coefficient or Kolmogorov-Smirnov test to test whether there is significant difference between the distributions of two features. Particularly, we can use principle component analysis to reduce the number of features needed on the labels.
Specific Aim 2: Develop new graph kernel methods for binding site prediction. The Co-PI will use the machine-learning approach to develop predictors for binding sites. The procedure includes (1) collect structures of complexes; (2) extract binding sites from the structures; (3) generate surface patches and graphs; (4) develop graph kernel methods for binding site prediction; and (5) evaluate the methods. Datasets collection Crystal structures of protein-protein complexes and protein-nucleic acid complexes will be collected from the PDB57. To ensure the quality of the dataset, we will discard structures with resolution value greater than 2.5 or R factor greater than 0.3. Many groups have tackled the problem of distinguishing biologically important contacts from crystal compact. Their results are deposited in databases like PQS58, PISA59, and PiQSi60. We will remove crystal contacts in the structures using these databases. The apo-state (unbounded) structures of the involving proteins will also be collected from the PDB if they are available. Extract binding sites Two types of definitions for binding-site residues have been widely used to extra binding sites from complex structures. One is based on the reduction of solvent accessible surface upon the formation of complex61. A residue is defined to be a binding-site residue if its solvent accessible surface is reduced by at least a certain amount during the formation of the complex. The second definition is based on the atom distance62. A residue is defined as a binding-site residue if its distance to the interacting partner is less than a certain distance. We will explore both types of definitions. Another issue in extracting binding site from PDB structure is that a protein may involve in multiple interactions, but a PDB structure may only shows one of them. Furthermore, some PDB structures only show a partial binding site due to incomplete structures63. Previously we have developed a tool, named TCBRP63 (http://bioweb.cs.ndsu.nodak.edu/Server/PPBindingprediction.html), for automatic extraction of complete binding sites from PDB complexes. We will use TCBRP for this step. Generate surface patches and graphs We will divide the protein surfaces into overlapping patches and represent them using the graph models developed in Specific Aim 1. For each residue on the protein surface, we will generate a patch that includes it and its contacting neighbors. In the training stage, a patch is assigned to either positive class (i.e., binding sites) or negative class (i.e., non-binding sites) depending on whether the center residue is a binding-site residue. In the prediction stage, the predicted class of a graph is assigned to its center residue.
A Graph Kernel Method for DNA-Binding Site Prediction Changhui Yan* and Yingfeng Wang CS, Utah State U, Logan, UT ABSTRACT: This paper presents a graph kernel method for predicting DNA-binding sites on protein structures. Surface patches are represented using labeled graphs. Then, the graph kernel method is used to calculate the similarities between graphs. A new surface patch is predicted to be interface or non-interface patch based on its similarities to known DNA-binding patches and non-DNA-binding patches. The proposed method achieves 88.7% accuracy, 89.7% specificity, and 87.7% sensitivity when tested on a representative set of 146 protein-DNA complexes using leave-one-out cross-validation. Then, the method is applied to identify DNA-binding sties on 13 unbound structures of DNA-binding proteins. In each of the unbound structure, the top 1 patch predicted by the proposed method precisely indicates the location of the DNA-binding site. Comparisons with other methods confirm the effectiveness of the method. Introduction Structural genomics projects are yielding an increasingly large number of protein structures with unknown function. As a result, computational methods for predicting functional sites on these structures are in urgent demand. There has been significant interest in developing computational methods for identifying amino acid residues that participate in protein-DNA interactions based on combinations of sequence, structure, evolutionary information, and chemical or physical properties. For example, Jones et al. (2003) analyzed residue patches on the surface of DNA-binding proteins and used electrostatic potentials of residues to predict DNA-binding sites. Later, they extended that method by including DNA-binding structural motifs (Shanahan, et al., 2004). In related studies, Tsuchiya et al. (2004) used a structure-based method to identify protein-DNA binding sites based on electrostatic potentials and surface shape, and Keil et al. (2004) trained a neural network classifier to identify patches likely to be DNA-binding sites based on physical and chemical properties of the patches. Neural network classifiers have also been used to identify protein-DNA interface residues based on a combination of sequence and structure information (Ahmad, et al., 2004). Recently, Tjong and Zhou (2007) developed a neural network method for predicting whether a surface residue is in the DNA-binding sites based on the sequence profile of that residue and its structural neighbors. On another track, several methods have been developed for predicting DNA-binding sites using only protein sequence-derived information as input (Ahmad and Sarai, 2005; Wang and Brown, 2006; Yan, et al., 2006). To date, the methods that take the advantage of structure-derived information achieve better results than those using only sequence-derived information. One common limitation of the above-mentioned methods is that the sequence and structural properties of a surface patch are input to machine-learning methods in the form of vectors. When the properties of a surface patch are encoded as a vector, the information of how these properties distribute over the surface is lost. For example, if a surface patch includes five amino acid residues, the above-mentioned methods will encode the amino acid identities of this surface patch as five independent values in a vector. In this representation, the spatial arrangement of these five residues on the surface patch is not encoded. Unfortunately, the spatial arrangement of properties on a surface patch plays a crucial role in determining the function of the surface patch. To overcome this limitation, this paper presents a graphical approach for DNA-binding site prediction. In this study, graphs are used to represent surface patches, such that the spatial arrangement of various properties on the surface is explicitly encoded. The similarities between surface patches are then computed using a graph kernel method. A voting strategy is then used to classify surface patches into DNA-binding sites versus non-binding sites. The proposed method achieves 88.7% accuracy, 89.7% specificity, and 87.7% sensitivity in leave-one-out cross-validation. When applied to set of unbound structures of DNA-binding proteins, the proposed method can precisely identify the locations of DNA-binding sites.
METHODS DNA-binding proteins DNA-binding proteins were obtained from our previous study (Yan, et al., 2006). In that study, we extracted all protein-DNA complexes from the PDB (Berman, et al., 2000). Then, the dataset was culled using PISCES (Wang and Dunbrack, 2003). The resulting dataset consisted of 171 proteins with mutual sequence identity ≤ 30% and each protein had at least 40 amino acid residues. All the structures have resolution better than 3.0 Å and R factor less than 0.3. In the current study, seven features are evaluated for their usefulness in the prediction of DNA-binding sites. Thus, seven features were calculated for each protein. Among them, structural conservation was calculated based on the alignment of structural neighbors (See details in section 2.2). 25 proteins were discarded because no structures neighbors were found. Finally, 146 DNA-binding proteins were used to evaluate the method in cross-validation. Features DNA was removed from the protein-DNA complexes and seven features were calculated for each amino acid of the protein: (1) Relative solvent accessibility was calculated using NACCESS (Hubbard, 1993); (2) Electrostatic potential was calculated using Delphi (Rocchia, et al., 2001) with the same parameters used in the study of Jones et a. (2003). The electrostatic potential of a residue is defined as the average of the electrostatic potentials at the locations of all its atoms as described in Jones et a. (2003); (3) Sequence entropy at each residue position (the sequence entropy for the corresponding column in the multiple sequence alignment) was extracted from the HSSP database (Sander and Schneider, 1991). Sequence entropy is a measure of sequence conservation. The lower the value, the more conserved is the corresponding residue position; (4) Surface curvature at each residue position was calculated using MSP (http://connolly.best.vwh.net/); (5) Pockets on protein surface were detected using Proshape (http://csb.stanford.edu/~koehl/ProShape/download.php). The pocket size of a residue is defined as the size of the pocket that the residue is located in. If a residue is not located in any pocket, then a value of 0 is assigned to the pocket size of the residue; (6) The DALI server (Holm and Sander, 1995) was used to search for structural neighbors in the PDB for each of the DNA-binding proteins. The DALI server returned a multiple alignment of the query structure and its structural neighbors. Then, structural conservation score was calculated for each residue position using Scorecons (Valdar, 2002) based on the multiple alignment; and (7) position-specific scoring matrix (PSSM) of a protein was built by running 4 iterations of PSI-BLAST (Altschul, et al., 1997) against the NCBI non-redundant (nr) database. In the PSSM, each residue position corresponds to 20 values. Thus, in total, each amino acid residue is associated with 26 attributes. All these attributes were normalized to the range of [0,1]. Interface residues and surface residues Interface residues are defined as in Jones et al. (2003). Solvent accessible surface area (ASA) was computed for each residue in the unbound protein (in absence of DNA) and in the protein-DNA complex. A residue is defined to be an interface residue if its ASA in the protein-DNA complex is less than its ASA in the unbound protein by at least 1Å2. A residue is defined to be a surface residue if its relative accessibility (i.e., ASA divided by overall surface area) in the unbound protein is >5%. Interface patches and non-interface patches For each DNA-binding protein, an interface patch and a non-interface patch were obtained. The interface patch included all the interface residues. The non-interface patch was randomly taken from the protein surface such that (1) it consisted of a group of contiguous surface residues; (2) it had the same number of residues as the interface patch; and (3) it did not include any interface residue. Graph representation of patches Each amino acid residue is represented using a node labeled with the 26 attributes of the residue. Two residues are considered contacting if the closest distance between their heavy atoms is less than the sum of the radii of the atoms plus 0.5 Å. An edge is added between two nodes if the corresponding residues are contacting. In this way, a surface patch of residues is represented as a labeled graph.
Graph kernel: Kernel methods are a popular method with broad applications in data mining. In a simple way, a kernel function can be considered as a positive definite matrix that measures the similarities between each pair of input data. It the currently study, a graph kernel method, namely shortest-path kernel, developed by Borgwart and Kriegel, is used to compute the similarities between graphs. The first step of the shortest-path kernel is to transform original graphs into shortest-path graphs. A shortest-path graph has the same nodes as its original graph, and between each pair of nodes, there is an edge labeled with the shortest distance between the two nodes in the original graph. In the current study, the edge label will be referred to as the weight of the edge. This transformation can be done using any algorithm that solves the all-pairs-shortest-paths problem. In the current study, the Floyd-Warshall algorithm was used. Let G1 and G2 be two original graphs. They are transformed into shortest-path graphs S1(V1, E1) and S2(V2, E2), where V1 and V2 are the sets of nodes in S1 and S2, and E1 and E2 are the sets of edges in S1 and S2. Then a kernel function is used to calculate the similarity between G1 and G2 by comparing all pairs of edges between S1 and S2. where, kedge( )is a kernel function for comparing two edges (including the node labels and the edge weight). Let e1 be the edge between nodes v1 and w1, and e2 be the edge between nodes v2 and w2. Then, where, knode( ) is a kernel function for comparing the labels of two nodes, and kweight( ) is a kernel function for comparing the weights of two edges. These two functions are defined as in Borgward et al.(2005): where, labels(v) returns the vector of attributes associated with node v. Note that Knode() is a Gaussian kernel function. was set to 72 by trying different values between 32 and 128 with increments of 2. where, weight(e) returns the weight of edge e. Kweight( ) is a Brownian bridge kernel that assigns the highest value to the edges that are identical in length. Constant c was set to 2 as in Borgward et al.(2005). Classification and cross-validation When the shortest-path graph kernel is used to compute similarities between graphs, the results are affected by the sizes of the graphs. Consider the case that graph G is compared with graphs Gx and Gy separately using the graph kernel: If Gx has more nodes than Gy does, then |Ex|>|Ey|, where Ex and Ey are the sets of edges in the shortest-path graphs of Gx and Gy. Therefore, the summation (i.e., SS( ) ) in K(G, Gx) includes more items than the summation in K(G, Gy) does. Each item (i.e., kedge( )) inside the summation has a non-negative value. The consequence is that if K(G, Gx)>K(G,Gy) it may not necessary indicate that Gx is more similar to G than Gy is, in stead, it could be an artifact of the fact that Gx has more nodes than Gy. To overcome this problem, a voting strategy is developed for predicting whether a graph (or a patch) is an interface patch: Algoritm Voting_Stategy (G) Input: graph G Output: G is an interface patch or non-interface patch Let T be the set of proteins in the training set Let v be the number of votes given to “G is an interface patch” v=0 While (T is not empty) { Take one protein (P) out of T Let Gint and Gnon-int be the interface and non-interface patches from P. If K(G, Gint)>K(G,Gnon-int), then increase v by 1 } If , then G is an interface patch Else G is a non-interface patch Using this strategy, when K(G, Gint) is compared with K(G, Gnon-int), Gint and Gnon-int are guaranteed to have identical number of nodes, since they are the interface and non-interface patches extracted from the same protein (see section 2.4 for details). Each time K(G, Gint)>K(G, Gnon-int) is true, one vote is given to “G is an interface patch”. In the end G is predicted to be an interface patch if “G is an interface patch” gets more than half of the total votes, i.e.,. Leave-one-out cross-validation was performed at protein level. In one round of the experiment, the interface patch and non-interface patch of a protein were held out as test examples. The remaining 145 proteins were used as training set.
results Identification of DNA-binding patches in leave-one-out cross-validation 146 interface patches and 146 non-interface patches were obtained from the dataset. The graph kernel method was used to compute similarities between patches and the voting strategy was used to classify these patches into interface versus non-interface patches. When evaluated using a leave-one-out cross-validation, this method achieves an overall accuracy of 88.7%. 87.7% (Sensitivity) of the interface patches and 89.7% (Specificity) of the non-interface patches were correctly predicted. Contributions of the features In the above experiment, all seven features were used to calculate similarities between graphs. To evaluate the importance of each feature, the leave-one-out cross-validation was repeated with only one feature being used at one time. Table 1 shows show that when only one feature is used, the method achieves the best performance (86.9% accuracy) with PSSM as input. When all seven features are used, the method achieves the highest accuracy (88.7%). We also tried all combinations of PSSM with one feature from the remaining. The results showed that adding one feature to PSSM only increased the accuracy by at most 0.1%. (To do combinations based on ranking of features) Table 1. Contributions of features Features PSSM1 E_P2 Ent3 StrCn4 rASA5 Cur6 Poc7 All8 Accuracy (%) 86.9 77.0 67.5 54.7 54.5 54.1 54.1 88.71 PSSM: position-specific scoring matrix; 2 E_P: electrostatic potential; 3 Ent: sequence entropy; 4 StrCn: structural conservation; 5 rASA: relative solvent accessibility; 6 Cur: surface curvature; 7 Poc: size of pocket where the residue is located; and 8 All: all 7 attributes were used. Predicting DNA-binding sites on unbound proteins 13 test proteins with both DNA-bound and unbound complexes in the PDB were taken from a previous study (Tjong and Zhou, 2007). 14 such proteins were considered in the study by Tjong and Zhou. Here, 2abk was discarded because the sequence identity between the bound and unbound proteins was only 45% (Tjong and Zhou, 2007). In this section, the DNA-binding sites on the 13 unbound proteins will be predicted using the graph kernel method. For each surface residue on the test proteins, we obtained a surface patch that included the residue and its 5 closest neighbors. Then, the patches were classified into interface versus non-interface patches using the 146 proteins as training set. For each test protein, the training set was filtered such that none of the proteins in the training set shares > 30% identical residues with the test proteins. For 8 of the 13 proteins (gray shading in Table 2), DALI was not able to find any structural neighbors in PDB. Thus, the structural conservation of these proteins could not be computed. For these 8 proteins, only PSSM was used to compute similarities in the graph kernel, since table 1 shows that the proposed method can still achieve high accuracy when only PSSM is used in the graph kernel. For the remaining 5 proteins, all seven features were used in the graph kernel. 3.3.1 The top 1 patch has significant overlaps with the actual DNA-binding site Using the voting strategy, each patch was assigned a # representing its votes. The higher the vote number, the more similar is the patch to interface patches. For each test protein, we sorted the patches based on the numbers of votes they get, such that the top 1 patch got the most votes and the last one got the least. Table 2 shows that on every test protein, the top 1 patch overlaps with the actual DNA-binding site. On 7 of the 13 proteins, all the six residues in the top 1 are actually interface resides (6 true positives, 0 false positive). When averaged over the 13 proteins, the top 1 patch contains 4.8 interface residues and 1.2 non-interface residues, i.e., on average, 80% of the residues in the top 1 patch are interface residues. These results show that on a test protein, the top 1 patch can precisely indicate the location of the actual DNA-binding site.
If a patch is randomly picked from a test protein, what is the probability (Prandom) to obtain a patch that is at least as good as the top 1 patch in terms of predicting the DNA-binding sites? For each test protein, Prandom is calculated as N/Nall , where Nall is the total number of patches on the protein, N is the number of the patches that have at least as many interface residues as the top 1 patch. The results (Table 2) show that for 9 of the 13 proteins, Prandom is less than 10%. The average Prandom for the 13 protein is 9.8%. This indicates the significance of the predicting method. Obtaining higher coverage by combining multiple top-ranking patches In the evaluation of DNA-binding site prediction methods, there are mainly two measures that researchers would be interested in: coverage (TP/Nint) and accuracy (TP/Npr), where TP is true positive, i.e. the number of residues that are predicted to be interface residues and are actually interface residues, Nint is the total number of interface residues and Npr is the number of residues that are predicted to be interface residues. Coverage shows percentage of the actual interface residues correctly predicted and accuracy is the percentage of the predicted interface residues that are actually interface residues. Fewer top-ranking patches are used More top-ranking patches are used The above section has shown that the top 1 patch can precisely indicate the location of the DNA-binding site on each test protein. However, since a patch has only 6 residues, the predictions solely based on the top 1 patch only have low coverage. We can obtain higher coverage by combining the predictions of multiple top-ranking patches. For example, if only top 1 patch is used to predict DNA-binding sites, the average coverage and accuracy are 23% and 81% for the 13 proteins. When the union of the top 3 patches is used to predict DNA-binding sites, coverage increases to 42%, but accuracy decreases to 72%. Figure 1 shows the tradeoff between coverage and accuracy when multiple top-ranking patches are used. Figure 1 shows the trend that as more top-ranking patches are used, coverage increases but accuracy decreases. If researchers prefer to identify more interface residues at the cost of lower accuracy, then they can choose to use more top-ranking patches to predict DNA-binding sites. The performance will fall at the right side of the curve. On the other hand, if they desire higher accuracy, then they can use fewer patches. Table 2. Predictions by the top 1patch Unbound Bound Top 1 patch PRandom(%)4 PDB id 1PDB id TP2 FP3 1iknA,C 1leiA,B 6 0 0.4 1g6nA,B 1zrfA,B 6 0 2.4 1zzkA 1zziA 6 0 2.9 1mml 1ztwA 4 2 4.2 1a2pC 1brnL 6 0 4.5 1ko9A 1m3qA 6 0 4.9 1qc9A 1cl8A,B 4 2 8.0 1lqc 1l1mA,B 6 0 8.7 1qzqA 1rfiB 2 4 9.5 1xx8A 1xyiA 6 0 10.7 1qqiA 1gxpA,B 4 2 19.3 1l3kA 1u1qA 3 3 25.6 2alcA 1f5eP 4 2 26.7 For the proteins in gray shading, the interfaces were predicted using only PSSM. For the others, all seven features were used; 2 TP: number of interface residues in the top 1 patch; 3 FP: number of non-interface residues in the top 1 patch. 4 Prandom: When a patch is randomly picked, the probability to obtaining a patch that contains at least as many interface residues as the top 1 patch.
Comparison with other methods Recently, Tjong and Zhou (2007) developed a neural network method for predicting DNA-binding sites. In their method, for each surface residue, the PSSM and solvent accessibilities of the residue and its 14 neighbors were used as input to a neural network in the form of vectors. In their publication, Tjong and Zhou showed that their method achieved better performance than other previously published methods. In the current study, the 13 test proteins were obtained from the study of Tjong and Zhou. Thus, we can compare the method proposed in the current study with Tjong and Zhou’s neural network method using the 13 proteins. Figure 1. Tradeoff between coverage and accuracy In their publication, Tjong and Zhou also used coverage and accuracy to evaluate the predictions. However, they defined accuracy using a loosened criterion of “true positive” such that if a predicted interface residue is within four nearest neighbors of an actual interface residue, then it is counted as a true positive. Here, in the comparison of the two methods, the strict definition of true positive is used, i.e., a predicted interface residue is counted as true positive only when it is a true interface residue. The original data were obtained from table 1 of Tjong and Zhou (2007), the accuracy for the neural network method was recalculated using this strict definition (Table 3). The coverage of the neural network was directly taken from Tjong and Zhou (2007). For each protein, Tjong and Zhou’s method reported one coverage and one accuracy. In contrast, the method proposed this study allows the users to tradeoff between coverage and accuracy based on their actual need. For the purpose of comparison, for each test protein, top-ranking patches are included into the set of predicted interface residues one by one in the decreasing order of ranks until coverage is the same as or higher than the coverage that the neural network method achieved on that protein. Then the coverage and accuracy of the two methods are compared. On a test protein, method A is better than B, if accuracy(A)>accuracy(B) and coverage (A)≥coverage(B). Table 3 shows that the graph kernel method proposed in this study achieves better results than the neural network method on 7 proteins (in bold font in table 3). On 4 proteins (shown in gray shading in table 3), the neural network method is better than the graph kernel method. On the remaining 2 proteins (in italic font in table 3), conclusions can be drawn because the two conditions, accuracy(A)>accuracy(B) and coverage (A)≥coverage(B), never become true at the same time, i.e., when coverage (graph kernel)>coverage(neural network), we have accuracy(graph kernel)<accuracy(neural network), but when coverage (graph kernel)<coverage(neural network), we have accuracy(graph kernel)>accuracy(neural network). Note that the coverage of the graph kernel method increases in a discontinuous fashion as we use more patches to predict DNA-binding sites. One these two proteins, we were not able to reach at a point where the two methods have identical coverage. Given these situations, we consider that the two methods tie on these 2 proteins. Thus, these comparisons show that the graph kernel method can achieves better results than the neural network on 7 of the 13 proteins (shown in bold font in Table 3). Additionally, on another 4 proteins (shown in Italic font in Table 3), the graph kernel method ties with the neural network method. When averaged over the 13 proteins, the coverage and accuracy for the graph kernel method are 59% and 64%. It is worth to point out that, in the current study, the predictions are made using the protein structures that are unbound with DNA. In contrast, the data we obtained from Tjong and Zhou’s study were obtained using proteins structures bound with DNA. In their study, Tjong and Zhou showed that when unbound structures were used, the average coverage decreased by 6.3% and average accuracy by 4.7% for the 14 proteins (but the data for each protein was not shown). In overall, the graph kernel outperforms the neural network method in majority cases. At the same time, we also notice the neural network method outperforms the graph kernel method on 4 proteins. This suggests that the two methods complement each other.
the method proposed in this study; 2 The method developed by Tjong and Zhou (2007); 3 Coverage=Ntp/Nint, where Ntp is the number of true positive and Nint is the total number of interface residues; 4 Accuracy=Ntp/Npr, where Ntp is the number of true positives and Npr is the number of residues predicted to be interface residues; 5. In their publication, Tjong and Zhou (2007) defined accuracy using a loosened criterion of “true positive” such that if a predicted interface residue is within four nearest neighbors of an actual interface residue, then it is counted as a true positive. Here, in the comparison of the two methods, the strict definition of true positive is used, i.e., a predicted interface residue is counted as true positive only when it is a true interface residue. As a result, the accuracy shown here are lower than those in table 1 of Tjong and Zhou (2007); 6On the proteins shown in bold font, the graph kernel method outperforms neural network method. On the proteins in gray shading, the neural network method outperforms the graph kernel method. The two methods tie on the remaining two proteins shown in italic font. Table 3. Comparison with other methods DISCUSSION This paper presents a graph kernel method for predicting DNA-binding sites on proteins. Cross-validation shows that the proposed method achieves 88.7% accuracy, 89.7% specificity, and 87.7% sensitivity. The effectiveness of the method is demonstrated by applying it to 13 unbound protein structures and comparing with other methods. Different from previous methods that represent sequence and structural properties of surface using vectors, the method proposed in this study uses labeled graphs. Compared to vectors, one advantage of labeled graphs is that they can specifically encode the spatial arrangement of the properties on protein surface. Since proteins and DNA interact in a 3-dimensional space, the spatial arrangement of the properties on protein surface plays a pivotal role in the interactions. Therefore, computational methods for prediction of the interface should consider the spatial arrangement of the properties. This paper presents a graph kernel method that can efficiently explore this information. ACKNOWLEDGEMENTS CY conceived of and designed the study, performed the computation and analysis, and drafted the manuscript. YW contributed partially to programming. All authors read and approved the final manuscript. REFERENCES Ahmad, S., Gromiha, M.M. and Sarai, A. (2004) Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information, Bioinformatics, 20, 477-486. Ahmad, S. and Sarai, A. (2005) PSSM-based prediction of DNA binding sites in proteins, BMC Bioinformatics, 6, 33. Altschul, S., Madden, T., Schaffer, A., Zhang, J., Zhang, Z., Miller, W. and Lipman, D. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucl. Acids Res., 25, 3389-3402. Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N. and Bourne, P.E. (2000) The Protein Data Bank, Nucl Acids Res, 28, 235-242. Borgwardt, K.M. and Kriegel, H.-P. (2005) Shortest-path kernels on graphs. The fifth IEEE International Conference on Data Minning (ICDM'05). 8. Borgwardt, K.M., Ong, C.S., Schonauer, S., Vishwanathan, S.V.N., Smola, A.J. and Kriegel, H.-P. (2005) Protein function prediction via graph kernels, Bioinformatics, 21, i47-56. Holm, L. and Sander, C. (1995) Dali: a network tool for protein structure comparison, Trends Biochem. Sci., 20, 478-480. Hubbard, S.J. (1993) NACCESS. Department of Biochemistry and Molecular Biology, University College, London. Jones, S., Shanahan, H.P., Berman, H.M. and Thornton, J.M. (2003) Using electrostatic potentials to predict DNA-binding sites on DNA-binding proteins, Nucl Acids Res, 31, 7189-7198. Keil, M., Exner, T. and Brickmann, J. (2004) Pattern recognition strategies for molecular surfaces: III. Binding site prediction with a neural network, J. Comput. Chem., 25, 779-789. Rocchia, W., Alexov, E. and Honig, B. (2001) Extending the applicability of the nonlinear Poisson-Boltzmann equation: Multiple dielectric constants and multivalent ions, J. Phys. Chem., B 105, 6507-6514. Sander, C. and Schneider, R. (1991) Database of homology derived protein structures and the structural meaning of sequence alignment, Proteins, 9, 56-68. Shanahan, H.P., Garcia, M.A., Jones, S. and Thornton, J.M. (2004) Identifying DNA-binding proteins using structural motifs and the electrostatic potential, Nucl Acids Res, 32, 4732-4741. Tjong, H. and Zhou, H.-X. (2007) DISPLAR: an accurate method for predicting DNA-binding sites on protein surfaces, Nucl. Acids Res., 35, 1465-1477. Tsuchiya, Y., Kinoshita, K.. (2004) Structure-based prediction of DNA-binding sites on proteins using the empirical preference of electrostatic potential and the shape of molecular surfaces, Proteins, 55, 885-894. Valdar, W.S.J. (2002) Scoring residue conservation, Proteins: Struct., Funct., Genet., 48, 227-241. Wang, G. and Dunbrack, R.L.J. (2003) PISCES: a protein sequence culling server, Bioinformatics, 19, 1589-1591. Wang, L. and Brown, S.J. (2006) BindN: a web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences, Nucl Acids Res, 34, W243-W248. Yan, C., Terribilini, M., Wu, F., Jernigan, R.L., Dobbs, D. and Honavar, V. (2006) Identifying amino acid residues involved in protein-DNA interactions from sequence, BMC Bioinformatics, 7, 262.
D≡mrmv. What if d points away from the intersection, , of the Cut-hyperplane (Cut-line in this 2-D case) and the d-line (as it does for class=V, where d = (mvmr)/|mvmr|? Then a is the negative of the distance shown (the angle is obtuse so its cosine is negative). But each vod is a larger negative number than a=(mr+mv)/2od, so we still want vod < ½(mv+mr) od d a d FAUST Obliqueformula:P(Xod)<aX any set of vectors (e.g., a training class). Let d = D/|D|. To separate rs from vs using means_midpoint as the cut-point, calculate a as follows: Viewing mr, mv as vectors ( e.g., mr ≡ originpt_mr ), a = ( mr+(mv-mr)/2 ) o d = (mr+mv)/2 o d r r r v v r mr r v v v r r v mv v r v v r v
PX o d < a PX o d < a = PdiXi<a = PdiXi<a Viewing mr, mv as vectors, a = ( mr + mv )o d stdr stdv stdr+stdv stdr+stdv FAUST Oblique vector of stdsD≡ mrmv, d=D/|D| To separate r from v: Using the vector of stds cutpoint , calculate a as follows: What are the purple stds? approach-1: for each coordinate (or dimension) calculate the stds of the coordinate values and for the vector of those stds. Let's remind ourselves that the formula given Md's formula, does not require looping through the X-values but requires only one AND program across the pTrees. r r r v v r mr rv v v r r v mv v r v v r v d
PXod<a = PdiXi<a pstdr pmr*pstdr + pmr*pstdv + pmv*pstdr - pmr*pstdr pstdr+pstdv pstdr +pstdv a = pmr + (pmv-pmr) = pmr*2pstdr + pmr*pstdv + pmv*2pstdr - pmr*2pstdr 2pstdr next? pmr + (pmv-pmr) = 2pstdr +pstdv pstdv+2pstdr By pmr, we mean this distance, mrod, which is also mean{rod|rR} FAUST Oblique D≡ mrmv, d=D/|D| Approach 2 To separate r from v: Using the stds of the projections , calculate a as follows: In this case the predicted classes will overlap (i.e., a given sample point may be assigned multiple classes) therefore we will have to order the class predictions. r r r v v r mrrv v v r r v mv v r v v r v r | v | r | d pmr | | r | r By pstdr, std{rod|rR} v | | r pmv | | v | v | v
R G ir1 ir2 std 8 15 13 9 1 8 13 13 19 2 5 7 7 6 3 6 8 8 7 4 6 12 13 13 5 5 8 9 7 7 R G ir1 ir2 mn 62.83 95.29 108.12 89.50 1 48.84 39.91 113.89 118.31 2 87.48 105.50 110.60 87.46 3 77.41 90.94 95.61 75.35 4 59.59 62.27 83.02 69.95 5 69.01 77.42 81.59 64.13 7 FAUST Satlog evaluation cls avg 4 2.12 2 2.36 5 4.03 7 4.12 1 4.71 3 5.27 above=(std+stdup)/gap below=(std+stddn)/gapdn suggest ord 425713 red green ir1 ir2 abv below abv below abv below abv below avg 1 4.33 2.10 5.29 2.16 1.68 8.09 13.11 0.94 4.71 2 1.30 1.12 6.07 0.94 2.36 3 1.09 2.16 8.09 6.07 1.07 13.11 5.27 4 1.31 1.09 1.18 5.29 1.67 1.68 3.70 1.07 2.12 5 1.30 4.33 1.12 1.32 15.37 1.67 3.43 3.70 4.03 7 2.10 1.31 1.32 1.18 15.37 3.43 4.12 pmr*pstdv + pmv*2pstdr 2pstdr a = pmr + (pmv-pmr) = pstdr +2pstdv pstdv+2pstdr NonOblique lev-0 1's 2's 3's 4's 5's 7's True Positives: 99 193 325 130 151 257 Class Totals-> 461 224 397 211 237 470 So the number of FPs is drastically reduced and TPs somewhat reduced. Is that better? If we parameterize the 2 (doubling) and adjust to max TPs and min FPs, what is the optimal multiplier parameter value? Next, low-to-high std elimination ordering. NonOblq lev-1 50% 1's 2's 3's 4's 5's 7's True Positives: 212 183 314 103 157 330 False Positives: 14 1 42 103 36 189 Oblique level-0 using midpoint of means 1's 2's 3's 4's 5's 7's True Positives: 322 199 344 145 174 353 False Positives: 28 3 80 171 107 74 Oblique level-0 using means and stds of projections (w/o cls elim) 1's 2's 3's 4's 5's 7's True Positives: 359 205 332 144 175 324 False Positives: 29 18 47 156 131 58 Oblique lev-0, means, stds of projections (w cls elim in 2345671 order)Note that none occurs 1's 2's 3's 4's 5's 7's True Positives: 359 205 332 144 175 324 False Positives: 29 18 47 156 131 58 Oblique level-0 using means and stds of projections, doubling pstd 1's 2's 3's 4's 5's 7's True Positives: 410 212 277 179 199 324 False Positives: 114 40 113 259 235 58 Oblique lev-0, means, stds of projs,doubling pstdr, classify, eliminate in 2,3,4,5,7,1 ord 1's 2's 3's 4's 5's 7's True Positives: 309 212 277 154 163 248 False Positives: 22 40 65 211 196 27 1 2 3 4 5 7 tot 359 205 332 144 175 324 1539 TP s1/(s1+s2) 29 18 47 156 131 58 439 FP 410 212 277 179 199 324 1601 TP 2s1/(2s1+s2) 114 40 113 259 235 58 819 FP no elim ord 309 212 277 154 163 248 1363 TP 2s1/(2s1+s2) 22 40 65 211 196 27 561 FP 234571 329 189 277 154 164 307 1420 TP 2s1/(2s1+s2) 25 1 113 211 121 33 504 FP 347512 355 189 277 154 164 307 1446 TP 2s1/(2s1+s2) 37 18 14 259 121 33 482 FP 425713 355 189 277 154 164 307 1446 TP s1/(s1+s2) 37 18 14 259 121 33 482 FP level1 50% Oblique lev-0, means,stds of projs,doubling pstdr, classify, elim 3,4,7,5,1,2 ord 1's 2's 3's 4's 5's 7's True Positives: 329 189 277 154 164 307 False Positives: 25 1 113 211 121 33 2s1/(2s1+s2) elim ord: 425713 TP: 355 205 224 179 172 307 FP: 37 18 14 259 121 33

















