Download
1 / 68
710 likes | 1.17k Views
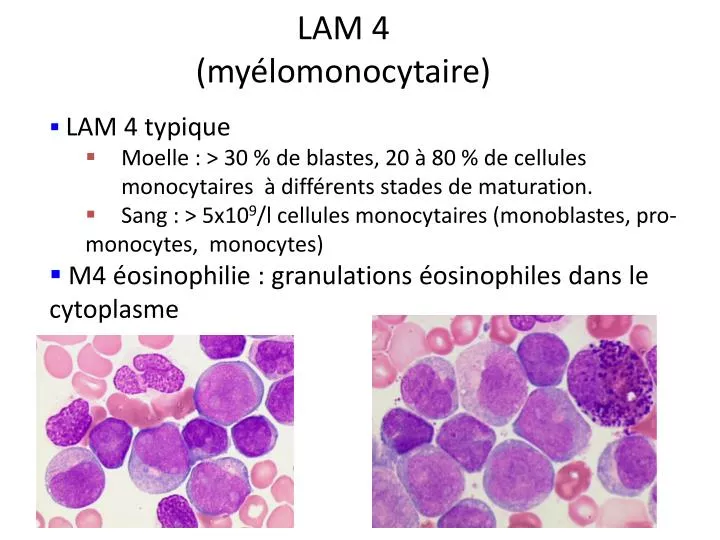
LAM 4 (myélomonocytaire). LAM 4 typique Moelle : > 30 % de blastes, 20 à 80 % de cellules monocytaires à différents stades de maturation. Sang : > 5x10 9 /l cellules monocytaires (monoblastes, pro-monocytes , monocytes) M4 éosinophilie : granulations éosinophiles dans le cytoplasme.

E N D
LAM 4 (myélomonocytaire) • LAM 4 typique • Moelle : > 30 % de blastes, 20 à 80 % de cellules monocytaires à différents stades de maturation. • Sang : > 5x109/l cellules monocytaires (monoblastes, pro-monocytes, monocytes) • M4 éosinophilie : granulations éosinophiles dans le cytoplasme
LAM 5 (monoblastique) • LAM5 a : peu différenciée ou monoblastique, plus de 80 % des cellules monocytaires de la moelle sont des monoblastes • LAM5b : différenciée, moins de 80 % des cellules de la moelle sont des monoblastes, présence de monocytes et promonocytes.
LAM 6 (Erythroleucémies) • Blastes > 30 % des éléments non érythroblastiques • Erythroblastes > 50 % des éléments nucléés avec dysérythropoïèse • Présence de micromégacaryocytes • Corps d ’Auer possibles • Evolution possible vers LAM1, LAM2, LAM4
LAM 7 (LA mégacaryocytaire) • Mégacaryoblastes • Reconnaissance par AC monoclonaux et/ou détection d ’activité péroxydasique en microscopie électronique
Syndrome de lyse tumorale (N. LEGUYADER) Neutropénie fébrile DEUX RISQUES MAJEURS
Deux risques majeurs • Syndrome de lyse tumoral • Forme hyperleucocytaire • Forme très tumorale • Risque: troubles ioniques, insuffisance rénale • Neutropénie fébrile • Chimio-induite • Infection: 1ère cause de mortalité chez le sujet neutropénique
Syndrome de lyse tumorale (N. LEGUYADER) Neutropénie fébrile DEUX RISQUES MAJEURS
Facteurs de risque • Masse tumorale élevée ou une hyperleucocytose • Taux de LDH élevé • Anomalies préexistantes de la fonction rénale pouvant être liée à la pathologie tumorale : - infiltration lymphomateuse ou leucémique - uropathie obstructive par compression ou infiltration tumorale
Pathologies et SLT • LNH de haut grade de malignité (lymphome de Burkitt +++) • LAL • LAM • décrit lors de neuroblastome, hépatoblastome, tératome sacrococcygien, rhabdomyosarcome...
Circonstances d ’apparition • Spontanément (Burkitt ++) • Hyperhydratation ++ • Chimiothérapie +++ • mais aussi hormonothérapie, radiothérapie, immunothérapie … • Dans les 24 - 48 premières heures de prise en charge et jusqu’à 5 jours suivant le début de la chimiothérapie
Cellule tumorale Anomalies biologiques isolées ou concomitantes Physiopathologie Nucléotides bases puriques LDH Phosphore Potassium Uricémie
Hyperuricémie • Anomalie la plus fréquente lors du SLT • Purines et précurseurs : métabolisme hépatique
Hyperuricémie • Sécrétion tubulaire acide urique taux plasmatique • Dans tube distal : [Urates] + pH + [sels] Diminution solubilité acide urique PRECIPITATION acide urique Néphropathie uratique
Hyperphosphorémie Hyperphosphaturie: par FG et réabsorption tubulaire (saturation transport) Hypocalcémie Précipitation phosphate de calcium Néphrocalcinoseaiguë Risque de convulsions +++
Prévention / Traitement = Hyperhydratation (3 l / m2 :G 5%, NaCl et Ca) sans potassium + / - Alcalinisation … + Maintien diurèse (furosémide : 0.5 à 1 mg/kg/6h) + Traitement hypo-uricémiant : - Allopurinol (Zyloric) + Epuration extra-rénale si troubles ioniques menaçants
Prévention / Traitement = Hyperhydratation (3 l / m2 :G 5%, NaCl et Ca) sans potassium + / - Alcalinisation + Maintien diurèse (furosémide : 0.5 à 1 mg/kg/6h) + Traitement hypo-uricémiant ou uricolytique : - Allopurinol (Zyloric) - Urate-oxydase
Syndrome de lyse tumorale Neutropénie fébrile DEUX RISQUES MAJEURS
Données épidémiologiques • L’infection est la première cause de mortalité chez les patients neutropéniques • Avant les années 70: septicémie à pyocyanique, 100 % de mortalité; 1969: 40 % avec carbenicilline et gentamicine • Aujourd’hui, risque de mortalité par infection dépend du statut de la pathologie sous jacente: < 5% à la phase initiale, mais très élevé en cas de pathologie réfractaire • Pronostic de certaines infections (aspergillose invasive…) reste très préoccupant
Données épidémiologiques • Facteurs de risques d’infections • Profondeur (< 500 PNN/mm3) et durée (> 7 jours) de la neutropénie; risque proche de 100 % si < 100 PNN/ mm3 pendant > 3 semaines; étude menée dans le service: chez patients ayant < 500 PNN, RR infection / à ceux ayant > 500 PNN est de 9,2 [3-28] • Lésions cutanéo-muqueuses chimio-induites • Cathéter central, et autres corps étrangers • Nutrition parentérale pour certains • Corticothérapie et ATB prolongée pour risque fongique, travaux pour aspergillose
Quels germes ? • Bactéries digestives • Bacilles gram négatifs • Bactéries cutanées: • Staphylocoques • Toujours y penser si cathéter central
Infections en aplasie: présentation clinique • Fièvre ++++ • Plusieurs définitions: 1 pic 38,5°C ou 38°C à 3 reprises en 24 h, actuellement, plutôt 1 pic 38,3°C ou 2 fois 38°C à 1 heure d’intervalle • Parfois le seul signe d’infection, mais peut manquer, en particulier en cas de corticothérapie +++ • N’est pas d’origine infectieuse dans environ 10 % des cas: néoplasique, chimiothérapie (ara C..), produits sanguins ou Ig, ampho B… • Documentation microbiologique dans 30% des cas, clinique dans 20% des cas, dite d’origine indéterminée mais possiblement infectieuse dans 40 % des cas
Infections en aplasie: examens de base • Examen clinique minutieux +++, peau, bouche et siège, signes de gravité (troubles hémodynamiques…) • Hémocultures (au mieux 2), technique rigoureuse, volume suffisant, anaérobie inutile sauf contexte particulier; frottis-goutte épaisse dans le contexte africain • CRP et/ou procalcitonine si disponible (Fleischhack et al, 2001, Erten et al 2004) • Compte de germe dans les selles (ou coproculture) • Radiographie de thorax • Selon contexte ou la disponibilité: prélèvements parasitologiques, bouche, gorge, peau, ECBU, virémie et Ag CMV, Ag aspergillaire, aspiration rhino-pharyngée pour virologie, PCR mycoplasme…LBA possible?
Infections en aplasie: conduite à tenir • URGENCE +++ • Antibiothérapie IV dans les 3 heures qui suivent l’apparition de la fièvre • Large spectre, rapidement bactéricide, association synergique • Tenir compte • ATCD infectieux de chaque patient • Signes cliniques (mucite et strepto, douleurs musculaires ou diarrhée et BGN…) • Évolution dans le temps de l’écologie microbienne chez les patients d’hémato-oncologie (Aquino et al, 1995) • Écologie de chaque service, et du profil de résistance des germes rencontrés
ßlactamine + aminoside +/- glycopeptide Évaluation à 48 H Syndrome infectieux persistant Apyrexie Poursuite traitement jusqu’à «sortie d’aplasie » Ajout glycopeptide Adaptation selon bactériologie Évaluation à 48-72 H Apyrexie Antifongique Reprisethermique Syndrome infectieux persistant Et /ou ajout glycopeptide, changement ATB
Infections en aplasie: conduite à tenir • Autresmesures • Aciclovirsimucite • Facteur de croissancehématopoïétique, en cas de syndrome septique non controlésidisponible
Infections en aplasie: conduite à tenir • Durée du traitement anti infectieux • Si trop court, risque de reprise du processusinfectieux, et de choc septique • Si trop long, risque de sélectionnergermerésistant, de favoriser infection fongique, surcoût important • Classiquementjusqu’à PNN > 500/mm3, voir plus tardsi infection documentée, maisarrêtprécoce possible (Katz et al 1993, Jones et al 1994…) pour population d’enfants en RC, apyrétiquedepuis > 24h, bon étatclinique, hémoculturesnégatives et amorce de sortie d’aplasie (plaquettes et/oumonocytes); économie 5000 $ / patient
Données générales Facteurs pronostiques Traitements Résultats actuels Leucémies aigues lymphoblastiques
Epidémiologie • Leucémies aiguës lymphoblastiques (LAL): 83% • Lignée B: 85% • Lignée T: 15% • Pic d’incidence entre 2 et 15 ans: LAL
Evolution du pronosticdes LAL de l’enfant Avant 1950 Durée médiane de survie = 3,4 mois 1950 - 1960 Quelques rémissions < 1 an 1960 2 % de survie à 7 ans 1970 20 % de survie à 7 ans 1980 60 % de survie à 7 ans 1990 75 % de survie à 7 ans
Epidémiologie • Actuellement: 80% de guérison • Survie variable selon les facteurs pronostiques • Age • Type B ou T • Leucocytose • Localisation: atteinte méningée ? • Réponse au traitement • Cytogénétique • Risque de rechute: standard / Haut risque • La stratégie thérapeutique joue un rôle pronostique • Plus un traitement est efficace, moins on retrouve les facteurs pronostiques.
Principes thérapeutiques des LAL • Adapter le traitement à chaque forme de LAL • Ne pas sur-traiter les formes à faible risque de rechute • Intensifier les formes à haut risque de rechute • Centres spécialisés • Optimisation des soins de support • Transfusion, Nutrition • Prévention des risques métaboliques • Traitement anti-infectieux • Abords veineux • Etudes multicentriques randomisées
Données générales Facteurs pronostiques Traitements Résultats actuels Leucémies aigues lymphoblastiques
Facteurs initiaux de pronostic défavorable Liés aux anomalies génétiques: Hypoploïdie < 45 chromosomes Translocation: t(9;22), t(4;11) Réarrangement du gène MLL Amplification 21q Transcrit de fusion: BCR-ABL MLL-AF4 • Liés au terrain: • Age: < 1 an, > 10 ans • Sexe: Garçon • Ethnique: race noire • Liés à la masse tumorale • GB > 50 000/mm3 (LAL B) • Syndrome tumoral important • Elargissement médiastinal • Atteinte du SNC • Liés au phénotype • Pro-B (CD19+, CD10-) • T
Facteurs pronostiques initiaux des LAL Smith M. et coll J Clin Oncol 1996, 14, 18-24
Anomalies cytogénétiques des LAL • Anomalies de nombre • Portant sur un seul chromosome: • perte (monosomie 7, 20, X) • gain (trisomie 21, X) • Bon pronostic: trisomie 4, 10, 17 • Portant sur plusieurs chromosomes • Hyperploïdie> 50, 30 % enfants, bon pronostic • Hypoploïdie< 46, 10 % LAL de l ’enfant, mauvais pronostic • Haploïdie : 26 à 28 chromosomes, 1 % LAL, mauvais pronostic • Pseudoploïdie: 46 chromosomes avec anomalie(s) inhabituelle(s), 35 % LAL, T ou B mature, pronostic intermédiaire • Anomalies de structures • Bon pronostic : t(12;21)/TEL-AML1 • Mauvais pronostic : t (9;22)/BCR-ABL, t(4;11)/MLL-AF4 • Autres : t(1;19); t(2;8), t(8;14), t(8;22) des LAL3
STRATIFICATION INITIALE: 4 grandes entités de LAL chez l ’enfant Nourrissons LAL-B LAL-BLAL-T < 1anRisque Haut Standard Risque 1-10 ans > 10 ans GB < 50.000 ou GB > 50.000 ou cytogénétique défavorable % 2-3% 55-60% 30% 12-15% Survie30-50% 85-90% 75-80% 70-80% à 5 ans STRATIFICATION SECONDAIREMENT AFFINEE PAR LA REPONSE PRECOCE AU TRAITEMENT
Facteurs pronostiques sous traitement • Corticosensibilité • < 1000 blastes / mm3 • à J8 d’un traitement par prednisone et 1 IT de méthotrexate • BFM 86 :
Facteurs pronostiques sous traitement • Chimiosensibilité • Myélogramme à J14 (ou J21), • M1: < 5 % blastes, M2: 5 à 25 % de blastes, • M3: > 25 % de blastes • Résultats Fralle 93 (LAL lignée B, âge > 1 an)
Apport de la génétique moléculaire en cours de traitement:Etude de la maladie résiduelle
Apport de la génétique moléculaire en cours de traitement • La persistance d’un niveau élevé de MRD est un facteur indépendant de mauvais pronostic (Cave et al, 1998)
Récapitulatif facteurs mauvais pronostiques CLINIQUE : Age <1 anou >10 ans, : Sexe masculin : Race noire : Syndrome tumoral important, élargissement médiastinal (+/-) : Atteinte du SNC (clinique et/ou LCR) HEMOGRAMME : GB > 50.000/mm3 (LAL de la lignée B seulement) IMMUNOPHENOTYPE : Immunophénotype pro-B (CD19+ CD10-) : Immunophénotype T (+/-) CYTOGENETIQUE : Hypodiploïdie < 45 chromosomes : Translocations défavorables : t(9;22), t(4;11) FISH : Réarrangement du gène MLL : Amplification 21q (iAMP21q) BIOLOGIE MOLECULAIRE: Transcrit de fusion BCR-ABL ou MLL-AF4 THERAPEUTIQUE : Chimiorésistance relative - cortico-résistance à J8 (> 1000 blastes / mm3 dans le sang après 7 jours de stéroïdes + IT MTX) - blastose médullaire > 5% à J14 ou J21 du traitement d’induction - maladie résiduelle élevée (> 1%) à J35-42 (immunologie ou biologie moléculaire) : Chimiorésistance avérée - blastose médullaire persistante à J35-42 (échec d’induction)
Données générales Facteurs pronostiques Traitements Résultats actuels Leucémies aigues lymphoblastiques
Traitementdes LAL : historique • 1948 : aminoptérine • 1950’-60’ : CT: 6-MP, VCR, MTX, • 1960’ : polychimiothérapie • 1960’-70 ’ : prophylaxie du SNC, • 1970’ : stratification, L-ASP, DNR • 1980’ : intensification décalée progrès du “supportive care” - 1990' : prise en compte de la réponse précoce - 2000’ : individualisation? désescalade ?
Traitement des LAL: principes • Stratification: selon les facteurs de risque initiaux et secondaires • 4 Phases thérapeutiques (2-3 ans) . INDUCTION . CONSOLIDATION . INTENSIFICATION(S) (rarement la greffe) . ENTRETIEN • Prophylaxie de l’atteinte du SNC
Traitement d ’induction • But : obtenir une rémission complète (RC) • Hémogramme normal (>1000 PNN; >100.000 plaquettes) • Myélogramme: < 5% de blastes, moelle de richesse normale • Modalités : • Préphase de corticoïdes • Vincristine (VCR) • Predniso(lo)ne (PRED) ou Dexamethasone (DXM) • Asparaginase (L-ASPA) • injections intrathécales (IT) 95% RC • Intérêt des anthracyclines : diminuer le risque de rechutes ultérieur des formes à haut risque.
Traitement de consolidation • Buts: • Maintenir la RC • Médicaments différents de ceux de l’induction • Pour éviter sélection de clones résistants • Traitements: • Aracytine, 6-MP, VP16, MTX +/- L-ASPA • Durée: 3 mois
Traitement d’intensification • Intérêt démontré par les études des groupes BFM et CCSG • Principe : • Séquence thérapeutique proche du traitement d'induction • 3 mois après la RC
Traitement d'entretien • But: éradiquer la maladie résiduelle • Modalités: • Administration continue • 6-MP (Purinethol) administrée tous les jours per os + Méthotrexatehebdomadaire (per os ou IM) • Adaptation dose: PNN entre 1000 et 1500 /mm3 • Réinductions mensuelles Vincristine + Prednisone + Intra-thécale • Durée optimale : 2 à 3 ans
Prophylaxie neuroméningée • Systématique • En l’absence de prophylaxie: Incidence des rechutes méningées 50 % (versus 2-3%) • Pourquoi ? • SNC: site sanctuaire pour les cellules leucémiques • Chimiothérapie ne passant pas correctement la barrière méningée • Modalités: • Injections intrathécales (MTX ± ARAC ± corticoïdes) • Méthotrexateà haute dose (3 à 8 g/m²) • Irradiation encéphale: forme à très haut risque • Atteinte méningée initiale • Présence d’une t(9;22) • Formes T hyperleucocytaires
