Download
1 / 1
10 likes | 164 Views
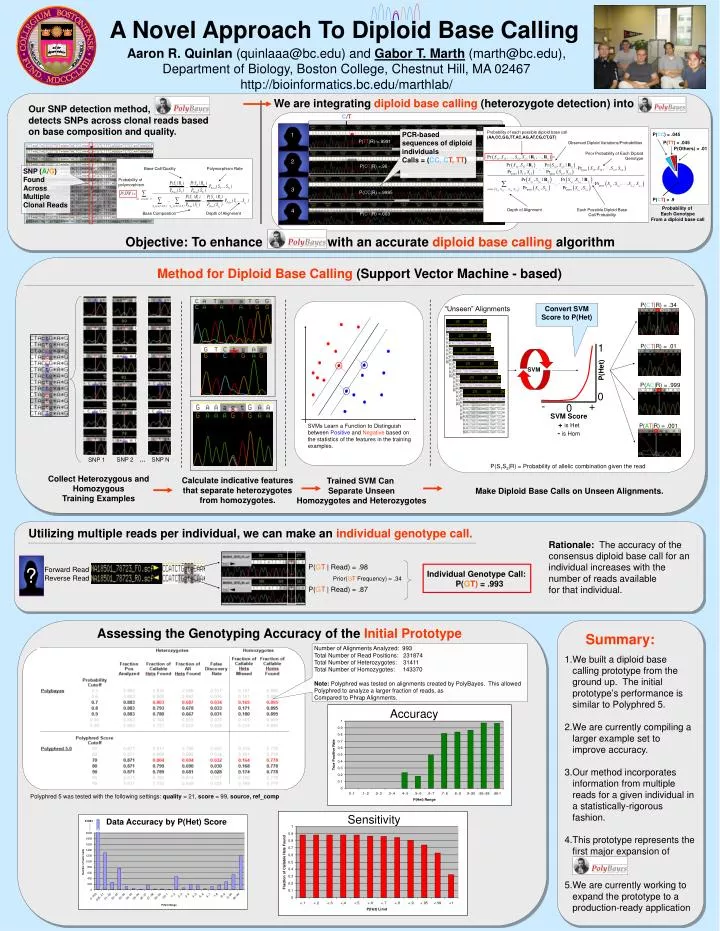
SVMs Learn a Function to Distinguish between Positive and Negative based on the statistics of the features in the training examples. A Novel Approach To Diploid Base Calling. Aaron R. Quinlan (quinlaaa@bc.edu) and Gabor T. Marth (marth@bc.edu),

E N D
SVMs Learn a Function to Distinguish between Positive and Negative based on the statistics of the features in the training examples. A Novel Approach To Diploid Base Calling Aaron R. Quinlan (quinlaaa@bc.edu) and Gabor T. Marth (marth@bc.edu), Department of Biology, Boston College, Chestnut Hill, MA 02467 http://bioinformatics.bc.edu/marthlab/ We are integrating diploid base calling (heterozygote detection) into Our SNP detection method, detects SNPs across clonal reads based on base composition and quality. C/T Probability of each possible diploid base call (AA,CC,GG,TT,AC,AG,AT,CG,CT,GT) PCR-based sequences of diploid individuals Calls = (CC, CT, TT) 1 P(CC) = .045 1 P(TT|R) =.9991 P(TT) = .045 Observed Diploid Variations/Probabilities P(Others) = .01 Prior Probability of Each Diploid Genotype 2 P(CT|R) =.96 Base Call/Quality Polymorphism Rate SNP (A/G) Found Across Multiple Clonal Reads Probability of polymorphism 3 P(CC|R) =.9995 P(CT) = .9 Probability of Each Genotype From a diploid base call 4 Depth of Alignment Each Possible Diploid Base Call/Probability Base Composition Depth of Alignment P(CT|R) =.003 Objective: To enhance with an accurate diploid base calling algorithm Method forDiploid Base Calling (Support Vector Machine - based) P(CT|R) = .34 “Unseen” Alignments Convert SVM Score to P(Het) 1 P(CT|R) =.01 P(Het) SVM P(AC|R) = .999 0 - + 0 SVM Score + is Het - is Hom P(AT|R) = .001 SNP 2 … SNP N SNP 1 P(S1S2|R) = Probability of allelic combination given the read Collect Heterozygous and Homozygous Training Examples Calculate indicative features that separate heterozygotes from homozygotes. Trained SVM Can Separate Unseen Homozygotes and Heterozygotes Make Diploid Base Calls on Unseen Alignments. Utilizing multiple reads per individual, we can make an individual genotype call. Rationale: The accuracy of the consensus diploid base call for an individual increases with the number of reads available for that individual. P(GT | Read) = .98 Assessing the Accuracy of the InitialPrototype: ? Forward Read Reverse Read Individual Genotype Call: P(GT) =.993 Prior(GT Frequency) = .34 P(GT | Read) = .87 Assessing the Genotyping Accuracy of the Initial Prototype Summary: Number of Alignments Analyzed: 993 Total Number of Read Positions: 231874 Total Number of Heterozygotes: 31411 Total Number of Homozygotes: 143370 Note: Polyphred was tested on alignments created by PolyBayes. This allowed Polyphred to analyze a larger fraction of reads, as Compared to Phrap Alignments. • We built a diploid base calling prototype from the ground up. The initial prototype’s performance is similar to Polyphred 5. • We are currently compiling a larger example set to improve accuracy. • Our method incorporates information from multiple reads for a given individual in a statistically-rigorous fashion. • This prototype represents the first major expansion of . • 5.We are currently working to expand the prototype to a production-ready application Accuracy Polyphred 5 was tested with the following settings: quality = 21, score = 99, source, ref_comp Sensitivity Data Accuracy by P(Het) Score 21851