Download

1 / 41
410 likes | 655 Views
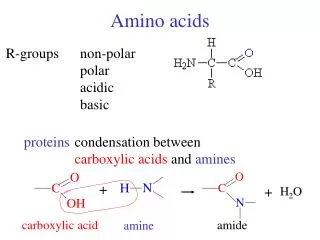
Amino Acids. Lecture 5. Introduction. Amino acids play central roles both as building blocks of proteins and as intermediates in metabolism. The 20 amino acids that are found within proteins convey a vast array of chemical versatility.

E N D
Amino Acids Lecture 5 M. Zaharna Clin. Chem. 2009
Introduction Amino acids play central roles both as building blocks of proteins and as intermediates in metabolism. The 20 amino acids that are found within proteins convey a vast array of chemical versatility. The precise amino acid content, and the sequence of those amino acids, of a specific protein, is determined by the sequence of the bases in the gene that encodes that protein. The chemical properties of the amino acids of proteins determine the biological activity of the protein. M. Zaharna Clin. Chem. 2009
Introduction In addition, proteins contain within their amino acid sequences the necessary information to: determine how that protein will fold into a three dimensional structure, and the stability of the resulting structure. It is important to keep in mind that one of the more important reasons to understand amino acid structure and properties is to be able to understand protein structure and properties. The vastly complex characteristics of even a small, relatively simple, protein are a composite of the properties of the amino acids which comprise the protein. M. Zaharna Clin. Chem. 2009
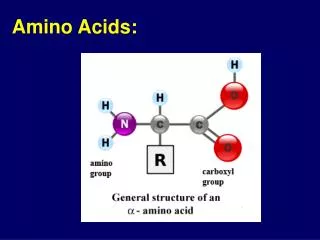
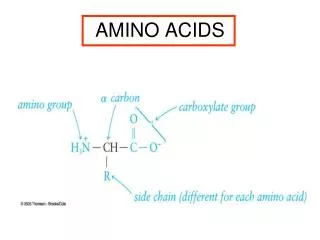
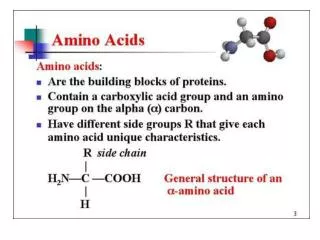
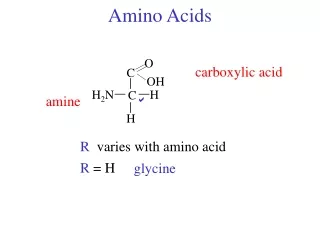
Amino Acid Structure An α-amino acid consists of: a central carbon atom, called the α carbon, linked to an amino group, a carboxylic acid group, a hydrogen atom, and a distinctive R group. The R group is often referred to as the side chain. The two mirror-image forms are called the L isomer and the d isomer. M. Zaharna Clin. Chem. 2009
Twenty kinds of side chains varying in size, shape, charge, hydrogen-bonding capacity, hydrophobic character, and chemical reactivity are commonly found. The remarkable range of functions mediated by proteins results from the diversity and versatility of these 20 building blocks. M. Zaharna Clin. Chem. 2009
Essential Amino Acids Humans can produce 11 of the 20 amino acids. arginine (often called semiessential as it is required for the young but not for The others must be supplied in the food. Failure to obtain enough of even 1 of the 9 essential amino acids, those that we cannot make, results in degradation of the body's proteins—muscle and so forth—to obtain the one amino acid that is needed. Unlike fat and starch, the human body does not store excess amino acids for later use—the amino acids must be in the food every day. M. Zaharna Clin. Chem. 2009
Metabolism • Proteolytic enzymes such as pepsin and trypsin digest dietary proteins into their constituent amino acids, the breakdown of body proteins is a source of amino acids. • The amino acid pool is used for: • the synthesis of body proteins • e.g. plasma, intracellular and structural proteins. • the synthesis of nonprotein nitrogen "NPN” containing compounds • such as purines, pyrimidines, creatine, histamine, thyroxine and others. • protein provides 12%-20% of the total daily body energy requirement M. Zaharna Clin. Chem. 2009
All tissues have some capability for synthesis of the non-essential amino acids, and conversion of non-amino acid carbon skeletons into amino acids. However, the liver is the major site of nitrogen metabolism in the body. In times of dietary surplus, the potentially toxic nitrogen of amino acids is eliminated via transaminations, deamination, and urea formation. The carbon skeletons are generally conserved as carbohydrate, via gluconeogenesis, or as fatty acid via fatty acid synthesis pathways. M. Zaharna Clin. Chem. 2009
In this respect amino acids fall into three categories: glucogenic, ketogenic, or glucogenic and ketogenic. Glucogenic amino acids are those that give rise to a net production of pyruvate or TCA cycle intermediates, such as α-ketoglutarate or oxaloacetate, (Alanine can be deaminated to pyruvate, arginine to – ketoglutarate ) Lysine and leucine are the only amino acids that are solely ketogenic, giving rise only to acetylCoA or acetoacetylCoA. M. Zaharna Clin. Chem. 2009
A small group of amino acids such as phenylalanine, and tyrosine give rise to both glucose and fatty acid precursors and are thus characterized as being glucogenic and ketogenic. M. Zaharna Clin. Chem. 2009
Aminoacidopathies They are rare inherited disorders of amino acid metabolism. Hereditary disorders of amino acid processing can be the result of : defects either in the breakdown of amino acids (activity of a specific enzyme ) or in the body's ability to get the amino acids into cells (membrane transport system ). More than 100 diseases have been identified that result from inborn errors of amino acid metabolism. Because these disorders produce symptoms early in life, newborns are routinely screened for several common ones. M. Zaharna Clin. Chem. 2009
Aminoacidopathies Phenylketonuria (PKU) Tyrosinemia Alkaptonuria Maple syrup urine disease Isovaleric Acidemia Homocystinuria Cystinuria M. Zaharna Clin. Chem. 2009
Phenylketonuria (PKU) Phenylalanine hydroxylase is an enzyme which converts phenylalanine to tyrosine. A deficiency of this enzyme leads to a buildup of phenylalanine which results in severe mental retardation. • Phenylalanine accumulates and is metabolized by alternate degradative pathway into phenylpyruvic acid and others leading to mental retardation. M. Zaharna Clin. Chem. 2009
This condition, known as phenylketonuria (PKU), is an autosomal recessive inborn error of metabolism. M. Zaharna Clin. Chem. 2009
Testing for PKU Testing is done either on the serum (Guthrie test) or on the urine. Testing is not valid until the newborn has ingested an ample amount of the amino acid phenylalanine, which is found in human and cow’s milk. Two or three days of intake are usually sufficient for the Guthrie test. Urine PKU testing is usually done after the infant is 4 to 6 weeks old. Normal blood phenylalanine level is about 1.2 - 3.4 mg/dl. In PKU, levels may range from 6 to 80mg/dl, usually greater than 30mg/dl M. Zaharna Clin. Chem. 2009
Guthrie bacterial inhibition assay Spores of B. Subtilis are incorporated into an agar plate that contains 2-thienylalanine, a metabolic antagonist to B. Subtilis growth. A filter paper disk impregnated with blood from the infant is placed on the agar. If the blood level of phenylalanine exceeds a range of 2 – 4 mg/dl, the phenylalanine counteracts the antagonist, and the bacterial growth occurs. M. Zaharna Clin. Chem. 2009
Guthrie bacterial inhibition assay • The infant must be at least 24h of age to ensure adequate time for enzyme and amino acid levels to develop. • The sample should be taken before the administration of antibiotics or transfusion of blood. • Premature infants can show false-positive results due to the immaturity of the liver's enzyme systems. M. Zaharna Clin. Chem. 2009
Microfluorometric assay For the direct measurement of phenylalanine in dried blood filter discs. It yields quantitative results, not affected by the presence of antibiotics. The procedure is based on the fluorescence of a complex formed of phenylalanine-ninhydrin- copper in the presence of a dipeptide "l-leucyl-l-alanine“. M. Zaharna Clin. Chem. 2009
Procedure extraction using trichloroacetic acid Then add a mixture of ninhydrin, succinate, leucylalanine and copper tartarate the fluorescence of the complex is measured using excitation/emission wavelengths of 360nm and 530nm respectively M. Zaharna Clin. Chem. 2009
Reference method The reference method for quantitative serum phenylalanine is HPLC. The normal limits of serum phenylalanine levels of full term normal weight newborns range from 1.2 mg/dl – 3.4 mg/dl. M. Zaharna Clin. Chem. 2009
Mass Spectrometry • Now, tandem mass spectrometry (MS/MS) is being used in screening for inherited disorders in newborns. Because both the increase in phenylalanine and the decrease in tyrosine levels seen in PKU can be identified, the ratio of phenylalanine to tyrosine (Phe/Try) can be calculated. • Using the ratio between metabolites rather than an individual level increases the specificity of the measurement and lowers the false-positive rate for PKU to less than 0.01%. • The MS/MS method has a greater sensitivity, detecting lower levels of phenylalanine and allowing for diagnosis of PKU as early as the first day of life. M. Zaharna Clin. Chem. 2009
Urine testing for phenylpyruvic acid Used for diagnosis in questionable cases and for monitoring of dietary therapy. It involves the reaction of ferric chloride with phenylpyruvic acid in urine to produce a green color. M. Zaharna Clin. Chem. 2009
Prenatal diagnosis and detection of carrier status In families with PKU, testing is now available using DNA analysis. The test is based on revealing multiple independent mutations at the phenylalanine hydroxylase locus. M. Zaharna Clin. Chem. 2009
Tyrosinemia Tyrosinemia is a genetic disorder characterized by elevated blood levels of the amino acid tyrosine, Tyrosine is a building block of most proteins. Tyrosinemia is caused by the shortage (deficiency) of one of the enzymes required for the multistep process that breaks down tyrosine. If untreated, tyrosine and its byproducts build up in tissues and organs, which leads to serious medical problems. M. Zaharna Clin. Chem. 2009
Tyrosinemia There are three types of tyrosinemia. Each has distinctive symptoms and is caused by the deficiency of a different enzyme. Type I tyrosinemia, the most severe form of this disorder, is caused by a shortage of the enzyme fumarylacetoacetate hydrolase. Type II tyrosinemia is caused by a deficiency of the enzyme tyrosine aminotransferase. Diagnostic criteria include an elevated tyrosine level using MS/MS coupled with a confirmatory test for an elevated level of the abnormal metabolite succinylacetone M. Zaharna Clin. Chem. 2009
Alkaptonuria It is due to the deficiency of homogentistate oxidase in the tyrosine catabolic pathway. Accumulation of homogentisic acid in urine causes its darkening upon exposure to a atmosphere. Alkaptonuric patients have no immediate problems, but later high levels of homogentisic acid gradually accumulate in connective tissue, causing generalized pigmentation of these tissues and an arthritis like degeneration. Urinalysis is done to test for alkaptonuria. When ferric chloride is added to the urine, it will turn the urine black in patients with alkaptonuria M. Zaharna Clin. Chem. 2009
Maple Syrup Urine Disease "MSUD" Maple syrup urine disease is an inherited disorder in which the body is unable to process certain amino acids properly. The condition gets its name from the distinctive sweet odor of affected infants' urine. Mutations in 4 genes cause maple syrup urine disease. These four genes provide instructions for making proteins that work together as a complex. The protein complex is essential for breaking down the amino acids leucine, isoleucine, and valine, which are present in many kinds of food M. Zaharna Clin. Chem. 2009
As a result, these amino acids and their byproducts build up in the body. Because high levels of these substances are toxic to the brain and other organs, their accumulation leads to the serious medical problems. M. Zaharna Clin. Chem. 2009
Tests A modified Guthrie test is used for neonatal screening. The metabolic inhibitor of B. Subtilis is 4-azaleucine. Positive test for MUSD, elevated level of leucine from a filter paper disc impregnated with infants blood will overcome the inhibitor and bacterial growth occurs. Confirmed diagnosis is based on finding increased levels of the three amino acids in plasma and urine with leucine being in highest concentration. A leucine level above 4 mg/dl is indicative of MUSD. MUSD can be diagnosed prenatally by measuring the decarboxylase enzyme concentration in cells cultured from amniotic fluid. MS/MS is also being used in testing for MSUD M. Zaharna Clin. Chem. 2009
Isovaleric Acidemia It results from a deficiency of the enzyme isovaleryl-COA dehydrogenase in the degradative pathway of leucine. The urine of newborns can be screened for isovaleric acidemia using MS/MS or chromotography M. Zaharna Clin. Chem. 2009
Homocystinuria . Homocysteine is an intermediate amino acid in the synthesis of cysteine from menthionine. Homocystinuria is caused by the impaired activity of the enzyme cystathionine -synthase which results in elevated plasma and urine levels of homocysteine and methionine. Newborns show no abnormalities, physical defects develop gradually with age. Clinical findings in late childhood include thrombosis, osteoporosis, dislocated eye lenses due to the lack of cysteine synthesis which is essential for collagen formation. M. Zaharna Clin. Chem. 2009
Neonatal screening with a Guthrie test using L-methionine sulfoximine as the metabolic inhibitor. Increased plasma methionine levels from affected infants will result in bacterial growth. Elevations in urinary homocystine can be detected by the cyanide-nitroprusside spot test. HPLC is the test used as the confirmatory method, with a methionine level greater than 2 mg/dL confirming positive results from the screening test. MS/MS is also used in screening programs to test for methionine levels. M. Zaharna Clin. Chem. 2009
Cystinuria • It is caused by a defect in the amino acid transport system rather than a metabolic enzyme deficiency. • Normally, amino acids are free filtered by the glomerulus and then actively reabsorbed in the proximal renal tubules. • In cystinuria, there is a 20-30 fold increase in the urinary excretion of cystine due to a genetic defect in the renal resorptive mechanism. • Because cystine is relatively insoluble, it tends to precipitate in the kidney tubules and form urinary calculi. • Cystinuria can be tested by cyanide-nitroprusside test. • Ion exchange chromatography can be used for quantitative analysis of amino acids in urine or plasma. M. Zaharna Clin. Chem. 2009
Amino Acid Analysis Blood samples drawn after 6-8 h fasting to avoid the effect of absorbed amino acids originating from dietary proteins. The sample is collected in heparin and the plasma is promptly removed from the cells "as cells contain higher concentration of amino acids", hemolysis should be avoided for the same reason. Deproteinization within 30 min of collection, analysis should be performed immediately or the sample should be stored at -20OC to -40OC. M. Zaharna Clin. Chem. 2009
Urinary amino acid analysis can be performed on a random specimen for screening purposes, but for quantitation, a 24 h urine preserved with thymol or organic solvent is required. Amniotic fluid also may be analyzed. The method of choice is the two-dimensional chromatography, the amino acids are allowed to migrate along one solvent front, and then the chromatogram is rotated 90O and a second solvent migration occurs. M. Zaharna Clin. Chem. 2009
The chromatogram is visualized by staining with ninhydrin, which gives a blue color with most amino acids. Confirmatory test for an amino acid disorder include separation and quantitation by cation-exchange chromatography using a gradient buffer elution. HPLC reversed-phase system equipped with fluorescence detection is another choice. Another technique that provides a highly specific and sensitive method for the measurement of amino acids is MS/MS M. Zaharna Clin. Chem. 2009