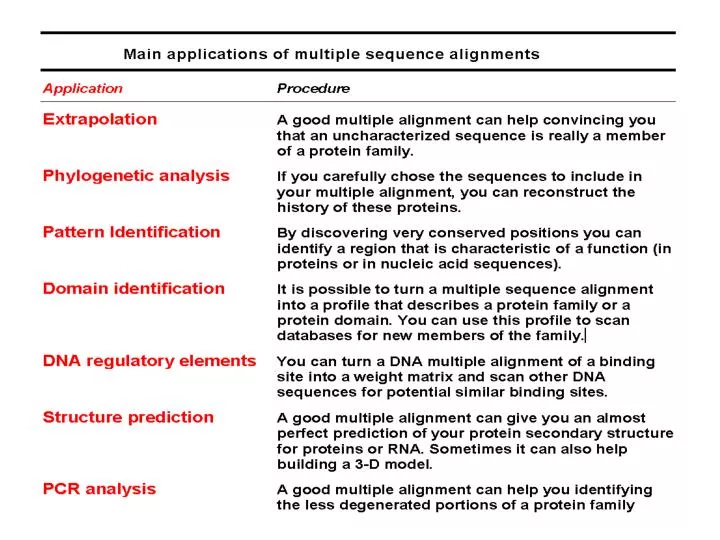
Download
1 / 28
370 likes | 918 Views
Multiple sequence alignment. Conserved blocks are recognized. Different degrees of similarity are marked. Multiple Sequence Alignment. VTISCTGSSSNIGAG-NHVKWYQQLPG VTISCTGTSSNIGS--ITVNWYQQLPG LRLSCSSSGFIFSS--YAMYWVRQAPG LSLTCTVSGTSFDD--YYSTWVRQPPG

E N D
Multiple sequence alignment Conserved blocks are recognized Different degrees of similarity are marked
Multiple Sequence Alignment VTISCTGSSSNIGAG-NHVKWYQQLPG VTISCTGTSSNIGS--ITVNWYQQLPG LRLSCSSSGFIFSS--YAMYWVRQAPG LSLTCTVSGTSFDD--YYSTWVRQPPG PEVTCVVVDVSHEDPQVKFNWYVDG-- ATLVCLISDFYPGA--VTVAWKADS-- AALGCLVKDYFPEP--VTVSWNSG--- VSLTCLVKGFYPSD--IAVEWESNG-- The purpose of multiple sequence alignments is to place homologous positions of homologous sequences into the same column.
ClustalW • Based on phylogenetic analysis • A phylogenetic tree is created using a pairwise distance matrix and nearest-neighbor algorithm • The most closely-related pairs of sequences are aligned using dynamic programming • Each of the alignments is analyzed and a profile of it is created • Alignment profiles are aligned progressively for a total alignment
Progressive multiple alignment • Perform pairwise alignments for all sequences Assume a match gives a score of 1, a mismatch is -0.25, indel is -0.5 1 -.25 1 1 1 1 Total Score: 4.75
Progressive multiple alignment • Create guide tree from pairwise alignments • Use tree to build multiple sequence alignment • Align most similar sequences first (give the most reliable alignments) • Align the profile to the next closest sequence • Align profiles to each other Multiple sequence alignment will be at the root of the tree
Operational options Output options Output options, matrix choice, gap opening penalty Gap penalties, output tree type File input in GCG, FASTA, EMBL, GenBank, Phylip, or several other formats Web ClustalW2 options:
Choose to run clustalw interactively or wait for results by email. Interactive may take some time so be patient
You can choose between a fast or full alignment. Full is more accurate and is what we will be using.
And this one We will use this option
Alignment - considerations • The programs simply try to maximize the number of matches • The “best” alignment may not be the correct biological one • Multiple alignments are done progressively • Such alignments get progressively worse as you add sequences • Mistakes that occur during alignment process are frozen in. • You will sometimes have to correct manually
Need more accuracy then Clustalw for low identity sequences?
Position Specific Iterated BLAST: PSI-BLAST The purpose of PSI-BLAST is to look deeper into the database for matches to your query protein sequence by employing a scoring matrix that is customized to your query.
PSI-BLAST is performed in five steps [1] Select a query and search it against a protein database – REGULAR BLAST [2] PSI-BLAST constructs a multiple sequence alignment then creates a “profile” or specialized position-specific scoring matrix (PSSM) – user-assisted – you can help choosing the candidates. [3] The PSSM is used as a query against the database [4] PSI-BLAST estimates statistical significance (E values) [5] Repeat steps [3] and [4] iteratively, typically 5 times. At each new search, a new profile is used as the query.
PSI-BLAST: self-positives PSI-BLAST is useful to detect weak but biologically meaningful relationships between proteins. The main source of false positives is the erroneous amplification of sequences not related to the query. For instance, a query with a coiled-coil motif may detect thousands of other proteins with this motif that are not homologous. Once even a single non-related protein is included in a PSI-BLAST search above threshold, it will not go away.
One way to check results: take newly found seqs and perform PSI-BLAST using them, then examine whether we ‘fish’ original seq (reciprocal identification)






















