Download

1 / 99

E N D
CELL CYCLE DNA REPAIR CELL DEATH
The 2001 Nobel Prize in Physiology or Medicine was awarded to Lee Hartwell, Paul Nurse, and Tim Hunt for their ground-breaking work on cell cycle regulation. Starting in the late 60s, Hartwell used budding yeast to identify mutants that blocked specific stages of cell cycle progression. Nurse, working in fission yeast in the 70s, went on to isolate mutants that could also speed up the cell cycle, thus focussing his attention on the original CDK kinase, cdc2. In the 80s, Hunt identified proteins in sea urchin extracts, the levels of which varied through the cell cycle hence "cyclins". All three have continued to make important advances in cell cycle research including the identification of checkpoints, mechanisms coupling cell morphology to the cell cycle, and identification of additional classes of kinases, cyclins, and inhibitors.
THE PHASES OF THE CELL CYCLE mitosis cytokinesis DNA replication
A CELL CYLE CONTROLLER SYSTEM COORDINATES THE CELL CYCLE MACHINERY
CHECKPOINTS MONITOR PROGRESSION TROUGH THE CELL CYCLE Are cell proliferative factors present?
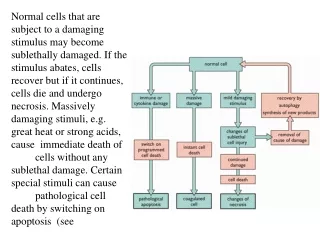
EXIT FROM THE CELL CYCLE-G0 Many times a cell will leave the cell cycle, temporarily or permanently. It exits the cycle at G1 and enters a stage designated G0 (G zero). A G0 cell is often called "quiescent", but that is probably more a reflection of the interests of the scientists studying the cell cycle than the cell itself. Many G0 cells are anything but quiescent. They are busy carrying out their functions in the organism. e.g., secretion, attacking pathogens. Often G0 cells are terminally differentiated: they will never reenter the cell cycle but instead will carry out their function in the organism until they die. For other cells, G0 can be followed by reentry into the cell cycle. Most of the lymphocytes in human blood are in G0. However, with proper stimulation, such as encountering the appropriate antigen, they can be stimulated to reenter the cell cycle (at G1) and proceed on to new rounds of alternating S phases and mitosis. G0 represents not simply the absence of signals for mitosis but an active repression of the genes needed for mitosis. Cancer cells cannot enter G0 and are destined to repeat the cell cycle indefinitely.
EXPERIMENTAL EVIDENCES OF CELL CYCLE REGULATORS Mixing nuclei together in the same cytoplasm (heterokaryon) to determine whether they could influence one another. CONCLUSION: THERE ARE DIFFUSIBLE FACTORS THAT CAN PROMOTE S OR M PHASE. THE S PHASE PROMOTING FACTOR (SPF) ONLY WORKS ON G1 NUCLEI. THE M PHASE PROMOTING FACTOR (MPF) WORKS ON EVERYTHING.
THE M PHASE PROMOTING FACTOR (MPF) -IDENTIFICATION OF CDK Xenopus laevis eggs:When a small sample taken from a meiosis -II metaphase arrested secondary oocyte is injected into a meiosis-I G2 phase primary oocyte, the G2-arrested cell will mature without progesterone and reach to metaphase of meiosis-II, because the secondary oocyte contains MPF. Cyclin dependent kinase (cdk) was isolated this way.
THE M PHASE PROMOTING FACTOR (MPF) -IDENTIFICATION OF CYCLIN Accumulation and degradation of cyclin B Sea urchin embryos. Found when looking at total proteins in a population undergoing synchronous division that some proteins go up and down with the cell cycle: cyclin.
THE M PHASE PROMOTING FACTOR (MPF) -IDENTIFICATION OF CYCLIN Sea urchin adult/embryo.
THE M PHASE PROMOTING FACTOR (MPF): CIKLIN+CDK Cdk 1 Cyclin B
THE M PHASE PROMOTING FACTOR (MPF): CIKLIN+CDK MPF IS ACTIVE ONLY AT HIGH CYCLIN CONCENTRATIONS
MPF IS SUFFICIENT TO INDUCE THE DRASTIC CHANGES OCCURING AT M-PHASE • chromosome condensation • breakdown of the nuclear envelope • reorganization of the actin cytoskeleton • reorganization of the microtubule cytoskeleton • assembly of the mitotic spindle • chromosome attachment to the kinetochore microtubules
Cdk4 Cdk2 Cdk1 D E A B(A) M G1 S G2 M G1 Start CDKs/CYCLINS OF HIGHER EUKARYOTES Cyclin-dependent kinases cyclins Cell cycle phases
Control of the Cell Cycle • The passage of a cell through the cell cycle is controlled by proteins in the cytoplasm. Among the main players in animal cells are: • CYCLINS • a G1 cyclin (cyclin D) • S-phase cyclins (cyclins E and A) • mitotic cyclins (cyclins B and A) • Their levels in the cell rise and fall with the stages of the cell cycle. • CYCLIN-DEPENDENT KINASES (CDKS) • a G1 Cdk (Cdk4) • an S-phase Cdk (Cdk2) • an M-phase Cdk (Cdk1) • Their levels in the cell remain fairly stable, but each must bind the appropriate cyclin (whose levels fluctuate) in order to be activated. They add phosphate groups to a variety of protein substrates that control processes in the cell cycle. • The anaphase-promoting complex (APC). (The APC is also called the cyclosome, and the complex is often designated as the APC/C.) The APC/C • triggers the events leading to destruction of the cohesins thus allowing the sister chromatids to separate; • degrades the mitotic cyclin B.
Steps in the cell cycle • A rising level of G1-cyclins bind to their Cdks and signal the cell to prepare the chromosomes for replication. • A rising level of S-phase promoting factor (SPF) — which includes cyclin A bound to Cdk2 — enters the nucleus and prepares the cell to duplicate its DNA (and its centrosomes). • As DNA replication continues, cyclin E is destroyed, and the level of mitotic cyclins begins to rise (in G2). • M-phase promoting factor (the complex of mitotic cyclins with the M-phase Cdk) initiates • assembly of the mitotic spindle • breakdown of the nuclear envelope • condensation of the chromosomes • These events take the cell to metaphase of mitosis. • At this point, the M-phase promoting factor activates the anaphase-promoting complex (APC/C) which • allows the sister chromatids at the metaphase plate to separate and move to the poles (= anaphase), completing mitosis; • destroys cyclin B. It does this by attaching it to the protein ubiquitin which targets it for destruction by proteasomes. • turns on synthesis of G1 cyclin for the next turn of the cycle; • degrades geminin, a protein that has kept the freshly-synthesized DNA in S phase from being re-replicated before mitosis. This is only one mechanism by which the cell ensures that every portion of its genome is copied once — and only once — during S phase.
MECHANISMS OF CDK REGULATION PHOSPHORILATION-DEPHOSPHORILATION
MECHANISMS OF CDK REGULATION INHIBITORY PROTEIN (P27/CKI) BINDING INACTIVATES THE CYCLIN-CDK COMPLEX
MECHANISMS OF CDK REGULATION UBIQUITIN-DEPENDENT PROTEIN DEGRADATION
THE G1/S CELL CYCLE CHECKPOINT DNA Proteinkinase activation Stable, Active p53 p53 foszforiláció p53 binds to the promoter of the p21 gene p53 degradation transcription P21 mRNS translation P21 (CKI) ACTIVE INACTIVE
THE G1/S CELL CYCLE CHECKPOINT The G1/S cell cycle checkpoint controls the passage of eukaryotic cells from the first “gap” phase (G1) into the DNA synthesis phase (S). Two cell cycle kinases, CDK4/6-cyclin D and CDK2-cyclin E, and the transcription complex that includes Rb and E2F are pivotal in controlling this checkpoint. During G1-phase, the Rb-HDAC repressor complex binds to the E2F-DP1 transcription factors, inhibiting downstream transcription. Phosphorylation of Rb by CDK4/6 and CDK2 dissociates the Rb-repressor complex, permitting transcription of S-phase-promoting genes including some that are required for DNA replication. Many different stimuli exert checkpoint control including TGFβ, DNA damage, contact inhibition, replicative senescence and growth factor withdrawal. The first four act by inducing members of the INK4 or Kip/Cip families of cyclin dependent kinase inhibitors (CKIs). TGFβ also inhibits the transcription of cdc25A, a phosphatase required for CDK activation. In response to DNA damage-induced activation of the ATM/ATR/Chk1/2 pathway, cdc25A is ubiquitinated and targeted for degradation via the SCF ubiquitin ligase complex. Targeted degradation of cdc25A in mitosis via the APC ubiquitin ligase complex allows progression through mitosis. Growth factor withdrawal activates GSK-3β, which phosphorylates cyclin D, leading to its rapid ubiquitination and proteosomal degradation. Ubiquitin/proteasome-dependent degradation and nuclear export are mechanisms commonly used to rapidly reduce the concentration of cell cycle control proteins. Some redundancy and tissue specific requirements exist as shown by animal models.
p53 • The p53 protein senses DNA damage and can halt progression of the cell cycle in G1. Both copies of the p53 gene must be mutated for this to fail so mutations in p53 are recessive, and p53 qualifies as a tumor suppressor gene. The p53 protein is also a key player in apoptosis, forcing "bad" cells to commit suicide. So if the cell has only mutant versions of the protein, it can live on — perhaps developing into a cancer. More than half of all human cancers have p53 mutations and have no functioning p53 protein. A genetically-engineered adenovirus can only replicate in human cells lacking p53. Thus it infects, replicates, and ultimately kills many types of cancer cells in vitro. Clinical trials are now proceeding to see if injections of this virus can shrink a variety of types of cancers in human patients. In some way, p53 seems to evaluate the extent of damage to DNA, at least for damage by radiation. At low levels of radiation, producing damage that can be repaired, p53 triggers arrest of the cell cycle until the damage is repaired. • At high levels of radiation, producing hopelessly damaged DNA, p53 triggers apoptosis. Possible mechanism: • Serious damage, e.g., double strand breaks(DSBs), causes a linker histone (H1) to be released from the chromatin. • H1 leaves the nucleus and enters the cytosol where • it triggers the release of cytochrome c from mitochondria leading to • apoptosis.
ATM (ataxia telangiectasia mutated) Symptoms of the disease ataxia telangiectasia. The ATM protein is involved in detecting DNA damage, especially double-strand breaks; interrupting (with the aid of p53) the cell cycle when damage is found; maintaining normal telomere length.
RB - the retinoblastoma gene • Retinoblastoma is a cancerous tumor of the retina. It occurs in two forms: • Familial retinoblastoma • Multiple tumors in the retinas of both eyes occurring in the first weeks of infancy. • Sporadic retinoblastoma • A single tumor appears in one eye sometime in early childhood before the retina is fully developed and mitosis in it ceases. • Familial retinoblastoma • Familial retinoblastoma occurs when the fetus inherits from both of its parents a chromosome (number 13) that has its RB locus deleted (or otherwise mutated). The normal Rb protein prevents mitosis. • Mechanism. The Rb protein prevents cells from entering S phase of the cell cycle. It does this by binding to a transcription factor called E2F. This prevents E2F from binding to the promoters of such proto-oncogenes as c-myc and c-fos. Transcription of c-myc and c-fos is needed for mitosis so blocking the transcription factor needed to turn on these genes prevents cell division. • Sporadic retinoblastoma • In this disease, both inherited RB genes are normal and a single cell must be so unlucky as to suffer a somatic mutation (often a deletion) in both in order to develop into a tumor. Such a double hit is an exceedingly improbable event, and so only rarely will such a tumor occur. (In both forms of the disease, the patient's life can be saved if the tumor(s) is detected soon enough and the affected eye(s) removed.)
THE G2/M CELL CYCLE CHECKPOINT A complex of checkpoint proteins recognizes unreplicated or damaged DNA and activates the protein kinase Chk1, which phosphorylates and inhibits the Cdc25 protein phosphatase. Inhibition of Cdc25 prevents dephosphorylation and activation of Cdc2.
THE G2/M CELL CYCLE CHECKPOINT The G2/M DNA damage checkpoint prevents the cell from entering mitosis (M-phase) if the genome is damaged. The cdk1-cyclin B complex is pivotal in regulating this transition. During G2-phase, cdk1 is maintained in an inactive state by the kinases Wee1 and Myt1. As cells approach M phase, the phosphatase cdc25 is activated by phosphorylation. Cdc25 then activates cdk1, establishing a feedback amplification loop that efficiently drives the cell into mitosis. DNA damage activates the DNA-PK/ATM/ATR kinases, initiating two parallel cascades that inactivate cdk1-cyclin B. The first cascade rapidly inhibits progression into mitosis: the Chk kinases phosphorylate and inactivate cdc25, which can no longer activate cdk1. The second cascade is slower. Phosphorylation of p53 dissociates it from MDM2, activating its DNA binding activity. Acetylation by p300/PCAF further activates its transcriptional activity. The genes that are turned on by p53 constitute effectors of this second cascade. They include 14-3-3, which binds to the phosphorylated cdk1-cyclin B complex and exports it from the nucleus; GADD45, which apparently binds to and dissociates the cdk1-cyclin B complex; and p21/Cip1, an inhibitor of a subset of the cyclin-dependent kinases including cdk1. cdk1
THE META/ANAPHASE TRANSITION-SPINDLE ASSEMBLY CHECKPOINT What if There Were No Checkpoint?
THE META/ANAPHASE TRANSITION-SPINDLE ASSEMBLY CHECKPOINT Unattached Kinetochores Cause a Checkpoint Delay
THE META/ANAPHASE TRANSITION-SPINDLE ASSEMBLY CHECKPOINT APC is the target of the spindle assembly checkpoint
THE META/ANAPHASE TRANSITION-SPINDLE ASSEMBLY CHECKPOINT Mad2 cycles through kinetochore and inhibits cdc20
MAD AND CANCER MAD (="mitotic arrest deficient") genes (there are two) encode proteins that bind to each kinetochore until a spindle fiber (one microtubule will do) attaches to it. If there is any failure to attach, MAD remains and blocks entry into anaphase. Mutations in MAD produce a defective protein and failure of the checkpoint. The cell finishes mitosis but produces daughter cells with too many or too few chromosomes (aneuploidy). Aneuploidy is one of the hallmarks of cancer cells suggesting that failure of the spindle checkpoint is a major step in the conversion of a normal cell into a cancerous one. Infection with the human T cell leukemia virus-1 (HTLV-1) leads to a cancer (ATL = "adult T cell leukemia") in about 5% of its victims. HTLV-1 encodes a protein, called Tax, that binds to MAD protein causing failure of the spindle checkpoint. The leukemic cells in these patients show many chromosome abnormalities including aneuploidy.
G2 G1 G1 M S CELL CYCLE CONTROL MECHANISMS Inappropiate environment DNA damage mitogen stimulation non replicated DNA Unattached kinetochores DNA damage CHECKPOINTS p53 Hct1 CKI APC G1-Cdk G1/S-Cdk S-Cdk Cdc25 M-Cdk KEY PLAYERS AND EVENTS Synthesis of G1/S cyclin Synthesis ofS cyclin DNA replication
SPONTANEOUS MUTATIONS The spontaneous mutation rate is low and different in different organisms. bacteria: 10-10-10-6/gene/generation The diploid nature of higher eukaryotes allows tolerance of higher mutation rates than in prokaryotes, provides greater genetic heterogeneity and thus evolutionary adaptability. Drosophila: 10-4-10-5/gene/generation Mouse: 10-5/gene/generation Human: 4x10-6-10-4/gene/generation Many of the recessive mutations are lethal: incompatible with life in homozygous condition. It is estimated that an average human is heterozygous for 3-5 recessive lethal mutations.
SPONTANEOUS MUTATIONS- TAUTOMERIC SHIFTS because of seldom and short lived tautomeric shifts, adenine and cytosine attain the rare imino, guanine and thymine the rare enol form. tautomeric shifts lead to unusual base pair formation (so-called mismatches)