Download

1 / 45
490 likes | 937 Views
PHT 415 BASIC PHARMACOKINETICS. Course Instructor: Prof. Dr. Hanaa Elsaghir Assistant lecturers: Text: Hand book of basic pharmacokinetics, Applied Biopharmaceutics and pharmacokinetics and lab notes

E N D
PHT 415BASIC PHARMACOKINETICS Course Instructor: Prof. Dr. Hanaa Elsaghir Assistant lecturers: Text: Hand book of basic pharmacokinetics, Applied Biopharmaceutics and pharmacokinetics and lab notes Grading: Quizzes (2.5 pt), Midterm (15 pt), Final (20 pt), Practical (10 pt), Attendance (2.5 pt) Lectures: Saturday and Monday (9-10 ) Office Hours: Sunday and Wednesday 11- 12 Email: helsaghir@ksu.edu.sa,hanaaelsaghir@yahoo.com
The student will be able to define: • Pharmacokinetics • Intravascular and extravascular administration • Absorption, disposition, distribution, metabolism, excretion, first-pass effect, enterohepatic cycling, compartment • The meaning of half- life, elimination rate constant, first order process, volume of distribution, clearance, renal clearance, and fraction excreted unchanged drug.
Estimate the values of half-life, elimination rate constant, volume of distribution, and renal clearance from plasma or blood concentrations of a drug following an IV dose • Estimate the values of half-life, elimination rate constant, and fraction excreted unchanged from urinary excretion data following an intravenous dose • Estimate the value of renal clearance of a drug from combined plasma and urine data . • Calculate the concentration of drug in the plasma and the amount of drug in the body with time following an intravenous dose, given values for the pharmacokinetic parameters.
Measurement of a drug in the body is limited usually to blood or plasma. • Blood receives drug from the site of administration as well as carries it to all the organs, including those in which the drug acts and those in which it is eliminated. • Sites of administration may be classified as either intravascular or extra vascular.
Intravascular administration refers to the placement of a drug directly into the blood, either intravenously or intra-arterially • Extravascular include the oral, sublingual, buccal, intramuscular, subcutaneous, dermal, pulmonary, and rectal routes • Drug administered extravascularly must be absorbed: no absorption step is required when a drug is administered intravascularly.
Once absorbed, a drug is distributed to the various organs of the body. Distribution is influenced by how well each organ is perfused with blood, organ size, binding of drug within blood and in tissues, and permeability of tissue membranes • The two principal organs of elimination, the liver and the kidneys • The kidneys are the primary site for excretion of the chemically unaltered, or unchanged, drug. • The liver is the usual organ for drug metabolism; however, the kidneys and other organs can also play an important metabolic role for certain drugs
The liver may also secrete unchanged drug into the bile. • The lungs are, or may be, an important route for eliminating volatile substances e.g. gaseous anesthetics. Absorption • Defined as the process by which unchanged drug proceeds from site of administration to site of measurement within the body. • Absorption is not restricted to oral administration. It occurs as well following IM, SC, and other extra vascular routes of administration
Monitoring intact drug in blood or plasma offers a useful means of assessing the entry of drug into the systemic circulation. Disposition • May be defined as, all processes that occur subsequent to the absorption of a drug. By definition, the components of disposition are distribution and elimination. • Elimination is the irreversible loss of drug from the site of measurement
Excretion is the irreversible loss of chemically unchanged drug. • Metabolism is the conversion of one chemical species to another... • Elimination occurs by two processes, excretion and metabolism. • Mass balance of a drug and related material with time in the body: • Dose= amount of drug at absorption site+ amount of drug in the body + amount of drug excreted +amount of metabolite in the body + amount of metabolite eliminated
Biopharmaceutics • Interrelationship of the physicochemical properties of the drug, dosage form in which the drug is given, and the route of administration on the rate and extent of systemic drug absorption Pharmacokinetics • Kinetics of drug absorption, distribution, and elimination (i.e. excretion and metabolism) • It involve experimental aspect and theoretical aspect
Measurement of drug concentrations • Sensitive, accurate, and precise analytical methods are available for the direct measurements of drugs in biologic samples, such as milk, plasma, and urine. Sampling of biologic specimens • Invasive methods such as sampling blood, spinal fluid , synovial fluid , tissue biopsy • Noninvasive methods include sampling of urine, saliva,feces, expired air,or any biologic material that can be obtained without parenteral or surgical intervention
Drug concentrations in blood, plasma, or serum • The most direct approach to assessing pharmacokinetics of the drug in the body is the measurement of drug concentration in thewhole blood, serum or plasma. Serum • Whole blood is allowed to clot and the serum is collected from the supernatant after centrifugation Plasma • Is obtained from supernatant of centrifuged whole blood to which an anticoagulant, such as heparin
Plasma level- time curve • Is generated by measuring the drug concentration in plasma samples taken at various time intervals after a drug product is administered • The concentration of drug in each plasma sample is plotted on rectangular coordinate graph against the corresponding time at which the plasma sample was removed. Drug concentrations in urine • Measurement of drug in urine is an indirect method to ascertain the bioavailability of a drug. The rate and extent of drug excreted in the urine reflects the rate and extent of systemic drug absorption.
Measurement of drug in feces may reflect drug that has not been absorbed after oral dose or may reflect drug that has been expelled by biliary secretion after systemic absorption. Significance of measuring plasma drug concentrations • allows for the adjustmen of the drug dosage in order to individualize and optimize therapeutic drug regimens • Guide to the progress of the diseased state and enable the investigator to modify the drug dosage accordingly. • Monitoring the concentration of drugs in the blood or plasma ascertains that the calculated dose actually delivers the plasma level required for therapeutic effect • Checking the plasma drug level is a responsive method of monitoring the course of therapy
Basic pharmacokinetics and pharmacokinetic model • Basic pharmacokinetics involves the quantitative study of various kinetic processes of drug disposition in the body • Drugs are in dynamic state within the body • A model is a hypothesis using mathematical terms to concisely describe quantitative relationships • Various mathematical models can be devised to simulate the rate processes of drug absorption, distribution, and elimination • The mathematical model makes possible the development of equations to describe drug concentrations in the body as function of time • A pharmacokinetic function relates an independent variable( time ) to a dependent variable (concentration of drug in plasma)
based on a set of time versus drug concentration data , a model equation is derived to predict the drug concentration in plasma with respect to time Pharmacokinetic models are used to: • predict plasma , tissue, and urine drug levels with any dosage form • calculate the optimum dosage regimen for each patient individually • correct drug concentrations with pharmacologic or toxicologic activity • evaluate differences in the rate or extend between formulations ( Bioequivalence ) • explain drug interactions
Experimental aspect involves, the development of biological sampling techniques, analytical methods for measurement of drugs and metabolites and procedures that facilitate data collection • Theoretical aspect involves the development of pharmacokinetic models that predict drug disposition after administration • Pharmacodynamics • Refer to the relationship between drug concentrations at the site of action ( receptor) and pharmacologic response including biochemical, and physiologic effect that influence the interaction of drug with receptor
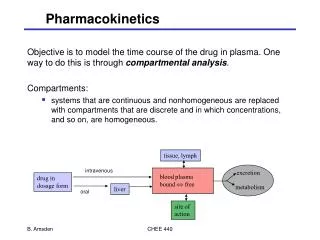
Pharmacokinetic Models • Compartment Models • mammillary model • caternary model • physiologic pharmacokinetic model( Flow model) Mammillary Model • The mammillary model is the most common compartment model used in pharmacokinetics • The model consist of one or more peripheral compartments connected to a central compartment • The central compartment represent plasma and highly perfused tissues which rapidly equilibrate with drug.
Mammillary model is a strongly connected system since one can estimate the amount of drug in any compartment of the system after drug is introduced into a given compartment • When an IV dose of drug is given, the drug enters directly in to the central compartment. • Elimination of drug occurs from the central compartment since the organs involved in drug elimination , kidney and liver, are well- perfused tissues. • Several types of compartment models are described in the slide
Drawing of the model has three functions • enables the pharmacokineticist to write differential equations to describe drug concentration changes in each compartment • gives visual representation of the rate process , and • shows how many pharmacokinetic constants are necessary to describe the process adequately.
PK and PD PK is the study of what the body does to a drug PD is the study of what a drug does to the body
Duration of Drug Therapy: Single Dose: such as to relieve a headache For the Rest of the Patient’s Life: such as in chronic diseases (epilepsy, diabetes) DRUG THERAPY
DOSAGE REGIMEN The manner in which a drug is taken is called a Dosage Regimen Duration of Drug Therapy and Dosage regimen will depend on the therapeutic objective. Objectivesof taking a drug are : Cure, Mitigation, Prevention
SUCCESSFUL DRUG THERAPY By optimally balancing the desirable and undesirable effects • Accurate diagnosis is made • Drug of choice • Knowledge of clinical state of patient • Understanding the pharmaco-therapeutic management of the disease • Answer the questions How much? How often? and How long?
Answers to Questions?????? How much? Recognizes that the magnitude of the therapeutic and toxic responses is a function of the dose given. How often? Recognizes the importance of time, in that the magnitude of the effect eventually declines with time following a single dose of drug. How long? Recognizes that a cost (in terms of side effects, toxicity, economics) is incurred with continuous drug administration.
In the Past Questions were answered by trial and error, i.e. the dose, interval between doses, and route of administration were selected and the patient’s progress was followed. This empirical approach established many dosage regimens but left many questions unanswered!!!!!! This empirical approach did not contribute toward establishing a safe, effective dosage regimen of another drug, i.e. did not help us understand how drugs work?
Event that follows drug administration Plasma conc of theophylline after an oral dose of 600-mg
Application of Pharmacokinetics Rarely is a drug placed at its site of action, most drugs are given orally and yet they act in the brain, heart, or elsewhere!!!! Which means a drug has to move from the site of administration to the site of action. Therefore to administer drugs optimally, knowledge is needed not only of the mechanisms of drug absorption, distribution, and elimination but also the kinetics of these processes, that is PK
Following Drug Administration 2 phases can be distinguished Pharmacokinetic Phase: in which the adjustable elements of dose, dosage form, frequency, and route of administration are related to drug level-time relationships in the body. Pharmacodynamic Phase: in which the conc of drug at the site of action(s) is related to the magnitude of the effect(s) produced.
Aimes of PK and PD Knowing the PK and PD of drugs will aid in designing a dosage regimen to achieve the therapeutic objective. OPTIMIZE PATIENT DRUG THERAPY BY MONITORING PK&PD RESPONSES
Advantages over the Empirical Approach 1- Distinction can be made between PK and PD causes of an unusual drug response. 2- Information gained about the PK of one drug can help in anticipating the PK of another drug. 3- Understanding the PK of a drug often explains the manner of its use. 4- Knowing the PK of a drug aids the clinician in determining the optimal dosage regimen for a patient and in predicting what may happen when a dosage regimen is changed.
Where to Measure Concentration? Rarely can the concentration of the drug at the site of action be measured directly; instead the concentration is measured at an alternative and more accessible site, the plasma.
What is then an optimal dosage regimen? It is the one that maintains the plasma concentration of a drug within the therapeutic window and then maintaining this concentration by replacing the amount of drug lost with time.
Initial and Maintenance Dose Regimen B Plasma conc Two different regimens A and B, B=2A Therapeutic Window Regimen A Time
Therapeutic Window (TW) The size and frequency of the maintenance dose depends on the width of the therapeutic window and the speed of drug elimination. When the window is narrow and the drug is eliminated rapidly, small doses must be given often to achieve therapeutic success. Both cyclosporine and digoxin have a narrow TW, but because cyclosporine is eliminated much more rapidly than digoxin, it has to be given…?…?…?…?…? ( more frequently or less frequently)
Oxytocin Oxytocin is an extreme example, it also has a narrow TW, but it is eliminated within minutes. The only means to ensure a therapeutic conc is to infuse it at a precise and constant rate directly into the blood (i.v. infusion). Oxytocin can not be given orally because it is destroyed in the GIT. Morphine can not also be given orally because it is extensively metabolized in the liver.
PROBLEM!!!Variability in Clinical Response Sources of variability: patient’s age, weight, degree of obesity, type and severity of disease, the patient’s genetic makeup, others drugs concurrently administered and environmental factors. Result: A standard dosage regimen of a drug may prove therapeutic in some patients, ineffective in others and toxic is still others.
Dosage Regimen Adjustment The need is greatest for drugs with narrow TW: Digoxin used to treat cardiac disorders. Phenytoin used to prevent epileptic convulsions Theophylline used to diminish chronic airway resistance in athmatics. Cyclosporine used as immunosuppressant in organ transplantation. Warfarin used as oral anti-coagulant.
Drug-Drug Interaction Interactions that result in a change in PK of a drug could be due to: Stimulation of drug metabolizing enzymes therefore increasing drug loss. Inhibition of drug metabolizing enzymes therefore slowing drug elimination and increasing its concentration in the blood. Interference with drug absorption.
SOLUTION A pragmatic approach to this problem would be to adjust the dosage until the desired objective is achieved. Control on a dosage basis alone, however, has proved difficult. Control is achieved more readily and accurately when plasma drug concentration data and the PK of the drug are known.
WHAT WE WILL DO IN THIS CLASS? Although the details of drug kinetics are complicated, it is fortunate that we can often approximate drug kinetic processes using “SIMPLE MATHEMATICAL MODELS”. The use of PK equations, rather than the derivation of the equations will be taught in this class.