Download

1 / 30
300 likes | 447 Views
Combinatorial Search Methods for Genotypes Associated with Lung Cancer Dumitru Brinza Department of Computer Science Georgia State University. Bioinformatics group. Dr. Alexander Zelikovsky Ph.D students: Jingwu He Weidong Mao Nisar Hundewale Dumitru Brinza Stefan Gremalschi

E N D
Combinatorial Search Methods for Genotypes Associated with Lung Cancer Dumitru Brinza Department of Computer Science Georgia State University
Bioinformatics group Dr. Alexander Zelikovsky Ph.D students: Jingwu He Weidong Mao Nisar Hundewale Dumitru Brinza Stefan Gremalschi Kelly Westbrooks
Collaboration with biologists and statisticians Dr. Andrey Perelygin, Georgia State University, Biology Department Dr. Liudmila Perelygina, Georgia State University, Biology Department Dr. Margo Brinton, Georgia State University, Biology Department Dr. Dragani Tommaso, National Institute of Cancer, Milan, Italy Dr. Robert Harrison, Georgia State University, Biology & CS Departments Dr. Art Vandenberg, Georgia State University, Department of Statistics Dr. Martin Fraser, Georgia State University, CS Department + Users of Our Software http://alla.cs.gsu.edu/~software
Research areas Genetics Genetic Epidemiology Computational Biology
Molecular biology terms Human Genome – all the genetic material in the chromosomes,length 3×109 base pairs Difference between any two people occur in 0.1% of genome SNP – single nucleotide polymorphism site where two or more different nucleotides occur in a large percentage of population. Genotype– The entire genetic identity of an individual, including alleles, SNPs, or gene forms. Haplotype – A single set of chromosomes (half of the full set of genetic material). Genotype is a mixture of two haplotypes.
Application domain We apply our methods to the gene data Usually datasets are given as a set of individuals each represented by genotype or pair of haplotypes Haplotypes: Wild type SNPs are notated as 0 Mutated SNPs are notated as 1 Genotypes: Homozygous SNPs are notated as 0,1 (mixture of 00,11) Heterozygous SNPs are notated as 2 (mixture of 01,10)
Focused problems Phasing (Haplotype inference) Tagging (Indexing) Disease association Disease susceptibility prediction
Phasing: Haplotype inference problem • Motivation: • Haplotype may contain large amount of genetic marker, which is responsible for human disease. • Great need in computational methods for extracting haplotype information from the given genotype information. • Inferring haplotypes from the genotypes is called the Phasing Problem: • Given:ngenotype vectors (0, 1 or 2), • Find: n pairs of haplotype vectors, one pair of haplotypes per • each genotype explaining genotypes • For individual genotype with h heterozygous sites there are 2h-1possible haplotype pairs explaining this genotype (h=20k for the genome-wide). also there are around 10% missing data. • This is hopeless without genetic model Motivation: Haplotype may contain large amount of genetic marker, which is responsible for human disease. Great need in computational methods for extracting haplotype information from the given genotype information. Inferring haplotypes from the genotypes is called the Phasing Problem: Given:ngenotype vectors (0, 1 or 2), Find: n pairs of haplotype vectors, one pair of haplotypes per each genotype explaining genotypes For individual genotype with h heterozygous sites there are 2h-1possible haplotype pairs explaining this genotype (h=20k for the genome-wide). also there are around 10% missing data. This is hopeless without genetic model
Phasing: The last achievement 2SNP – one of the best phasing method (scalable and accurate). Can phase with a high accuracy 50 genotypes with 10k SNPs in one minute versus other methods 1k in 1 month and less accurate. Journals: TCBB 2006, Phasing of 2-SNP Genotypes based on Non-Random Mating Model, to appear BIOINFORMATICS 2006, 2SNP: Scalable Phasing Based on 2-SNP Haplotypes IJBRA 2005, Family Trio Phasing and Missing Data Recovery Refereed conferences and posters: IWBRA 2006, Phasing of 2-SNP Genotypes based on Non-Random Mating Model GTICB 2005, 2SNP: New Scalable Phasing Method RECOMB 2005, Family Trio Phasing and Missing Data Recovery IWBRA 2005, Phasing and Missing Data Recovery in Family Trios WABI 2004, Linear Reduction for Haplotype Inference • Collect statistics on haplotype/genotype frequencies for any 2 SNPs • For each 2 SNPs compute weights reflecting likelihood of trans-/cis- • For each genotype g: • Find Maximum Spanning Tree for the complete graph G(g ) where vertices are heterozygous sites • Color G(g ) vertices and phase based on colors • Missing data recovery • Recover each missing site based on the closest haplotype with the phased site
Tagging (Indexing) Motivation: Human genome consists of 3×107 base pairs. It is desirable to reduce the number of SNPs that should be analyzed to a small number of informative representatives called Tag SNPs (Indexing SNPs). Tagging Problem: Extracting informative SNPs from the genotype/haplotype data Given: a sample S of population P of genotypes each with m SNPs Find: positions of k (k < m) tag SNPs such that one can reconstruct an entire genotype g in P from its restriction g' on k tag SNPs How to choose the right tags?
Tagging: The last achievement MLR-Tagging– Tag SNP Selection Based on Unphased Genotype data Using Multivariate Least Square Prediction. For a given k, it choose TAGs which represent initial dataset better than TAGs chosen by another methods. Journals: NanoBioscience 2006, Tag SNP Selection Based on Multivariate Linear Regression, to appear BIOINFORMATICS 2006, Tag SNP Selection Based on Multivariate Linear Regression, to appear IJBRA 2005, Linear Reduction Methods for Tag SNP Selection Refereed conferences and posters: EMBC 2006, Multiple Linear Regression for Index SNP Selection on Unphased Genotypes, submitted GRC 2006, Haplotype Tagging using Support Vector Machines RECOMB 2005, Linear Independent Tagging of Genotypes EMBC 2004, Linear Reduction Methods for Tag SNP Selection • Stepwise Index SNP Algorithm: • Choose as a tag the SNP which best predicts all other SNPs • Choose the next one which together with a first best predicts all other SNPs and so on. • Prediction method = MLR:
Genetic Epidemiology Genetic epidemiology - searching for genetic risk factors for diseases. Monogenic diseases A mutated gene is entirely responsible for the disease Typically rare in population: < 0.1% Complex disease Affected by the interaction of multiple genes Significance of risk factor is measured by risk rate or odds ratio.
Disease association Motivation: Analysis of variation in suspected genes in disease and nondisease individuals is aimed at identifying SNPs with considerably higher frequencies among the disease individuals than among the nondisease individuals. Most searches are done on a SNP-by-SNP basis. Common diseases can be caused by combinations of several unlinked gene variations We address the computational challenge of searching for such multi-gene causal combinations Disease association analysis searches for a SNPs or multi-SNP combinations with frequency among disease individuals considerably higher than among nondisease individuals. The number of multi-SNP combinations is infeasible high (3100 for 100 SNPs). How to find associated multi-SNP combinations without total checking?
Statistical significance Multi-SNP combination (MSC) define a set of disease and nondisese individuals MSC is considered statistically significant if the frequency of disease and nondisese distribution has p-value < 0.05 A lot of reported findings are frequently not reproducible on different populations. It is believed that this happens because the p-values are unadjusted to multiple testing – indeed, if the reported SNP is found among 100 SNPs then the probability that the SNP is associated with a disease by mere chance becomes roughly 100 times larger (Bonferroni). Multiple testing adjusted p-value: Bonferroni is too crude (e.g., 3-SNP combinations among 100 SNPs, p < 0.05×10-6) We adjust resulted p-values via randomization In our search we report only MSC with adjusted p-value < 0.05
Disease-Associated Multi-SNP Combinations Search Problem Formulation: Given a population of n genotypes (or haplotypes) each containing values of m SNPs and disease status (diseased or nondisease) Find all multi-SNP combinations with multiple testing adjusted p-value of the frequency distribution below 0.05.
Proposed Methods Exhaustive Search (ES): In order to find a multi-SNP combination with the p-value of the frequency distribution below 0.05, we should check all one-SNP, two-SNP, ..., m-SNP combinations. Complete searching is infeasible even for small number of SNPs We restrict searching to 1,2,3,4,5 SNPs Indexed Exhaustive Search (IES): Exhaustive search on the indexed datasets obtained by extracting k indexed SNPs with MLR based tagging method. Can perform complete searching for the larger datasets For wide-genome study number of tags can’t be reduced to 5-10 tags. Therefore, IES will not be able to perform complete search
Proposed Methods Definition: Disease-closure of a multi-SNP combination C is a multi-SNP combination C’, with maximum number of SNPs, which consists of the same set of disease individuals and minimum number of nondisease individuals. Combinatorial Search (CS): Similar to ES check all one-SNP, two-SNP, ..., m-SNP disease-closed combinations. Disease-closure allow finding of the statistically significant MSC on the earlier stage of searching. Trivial MSCs and MSCs which coincide after disease-closure are avoided. That significantly speedups the searching. Faster than ES Finds more significant association on the early stage of searching Still slow for wide-genome studies Indexed Combinatorial Search (ICS): Combinatorial search on the indexed datasets obtained by extracting k indexed SNPs with MLR based tagging method. Can perform complete searching for the larger datasets
Results for Disease Association Datasets: Crohn's disease (Daly et al ): inflammatory bowel disease (IBD). Location: 5q31 Number of SNPs: 103 Population Size: 387 case: 144 control: 243 Autoimmune disorders (Ueda et al) : Location: containing gene CD28, CTLA4 and ICONS Number of SNPs: 108 Population Size: 1024 case: 378 control: 646 Tick-borne encephalitis dataset of (Barkash et ) : Location: containing gene TLR3, PKR, OAS1, OAS2, and OAS3. Number of SNPs: 41 Population Size: 75 case: 21 control: 54 CS and ICS find statistically significant MSCs while no any statistically significant MSCs where found before.
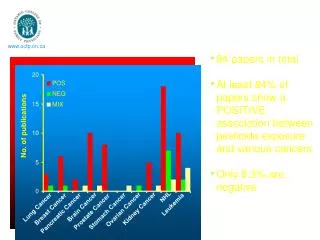
Disease Association for Lung Cancer Dataset of Lung Cancer disorder Provided by Dragani Tomasso, National Institute of Cancer, Milan 250 disease individuals and 300 nondisease individuals represented by their genotypes with 170 SNPs each. 7 statistically significant multi-SNP combinations were found Now the obtained MSC are checked on the larger population for the results replication. Preliminary results can be found in: Spinola, M., Meyer, P., Kammerer, S. et al. (2006) Association of the PDCD5 Locus With Lung Cancer Risk and Prognosis in Smokers, American Journal of Clinical Oncology, 24:11.
Published & to publish Results Brinza, D., and Zelikovsky, A. (2006) Combinatorial Methods for Disease AssociationSearch and Susceptibility Prediction, 6th Workshop on Algorithms in Bioinformatics(WABI'06),submitted Brinza, D., He, J., and Zelikovsky, A. (2006) Combinatorial Search Methods for Multi-SNP Disease Association, International Conference of the IEEE Engineering In Medicine and Biology Society (EMBC'06),submitted Brinza, D. and Zelikovsky, A. (2006) Search for multi-SNP Disease Association, The Fifth International Conference on Bioinformatics of Genome Regulation and Structure (BGRS'06), to appear Barkhash, A., Perelygin, A., Brinza, D., Pilipenko, P., Bogdanova, YU., Romaschenko, A., Voevoda, M. and Brinton, M. (2006) GENETIC RESISTANCE TO FLAVIVIRUSES, The Fifth International Conference on Bioinformatics of Genome Regulation and Structure (BGRS'06), to appear Barkhash, A., Perelygin, A., Brinza, D., Pilipenko, P., Bogdanova, YU., Romaschenko, A., Voevoda, M. and Brinton, M. (2006) VARIABILITY IN THE 2’-5’ OLIGOADENYLATE SYNTHETASE (OAS) GENE CLUSTER IS ASSOCIATED WITH SEVERITY OF TICK-BORNE ENCEPHALITIS VIRUS-INDUCED DISEASE IN RUSSIAN PATIENTS, 9-th Southeastern Regional Virology Conference 2006
Disease susceptibility prediction • Motivation: • There is a great need of making a diagnosis based on an individual’s DNA • Disease susceptibility: Predict a disease status of an individual based on its genotype/haplotypes • Given:Genotypes of disease and nondisease individuals, and • genotype of a testing individual • Find: The disease status of the tested individual • The existing methods for susceptibility prediction are very inaccurate! • (62%-70% prediction rate, when 50% gives random guessing) • We address the problem of developing very accurate and reliable methods for disease susceptibility prediction based on gene data. Refereed conferences and posters: WABI 2006, Combinatorial Methods for Disease AssociationSearch and Susceptibility Prediction GrC 2006, Genotype Susceptibility and Integrated Risk Factors for Complex Diseases EMBC 2005, A Combinatorial Method for Predicting Genetic Susceptibility to Complex Diseases RECOMB 2005, Graph Methods for Inheritated Disease Prediction
Complimentary Greedy Search based Prediction We assume existence of two types of MSCs: Risk and Resistance factors Risk MSCs dominate in disease individuals Resistance MCSs dominate in nondisease individuals Formulate disease association problem as an optimization problem of finding the best risk MSC and the best resistance MSC. Use a greedy algorithm with fast runtime which finds the MSCs enough close to the optimum Clustering: For the given training dataset find the best risk MSC and remove all people which are defined by this MSC. Find the next best MSC and remove all people defined by second MSC. Repeat that procedure until all disease individuals are covered by some MSC. We will obtain risk MSC cover (clustering) of all individuals Similarly obtain resistance MSC cover (clustering)
Clustering-based Model-Fitting Prediction Algorithm For a given individual with unknown status assume that it is disease individual and insert it in the training dataset, Sd. Obtain risk and resistance clustering of Sd. For each genotype obtain a ratio of PPV of its risk cluster over PPV of its resistance cluster (risk cluster cover not only disease individuals) Find a tradeoff between ratios which are considered diseased and ratios which are considered undiseased with objective of minimizing the error Check the ratio of inserted individual, and take a decision about its status. Repeat the same procedure over set Sn where unknown individual is introduced as nondisese individual If decision of both procedures coincide then take a final decision If not then take a decision of the procedure which error is less
LOO Cross Validation Results Leave-one-out cross validation results of four prediction methods for three real data sets. Results of combinatorial search-based prediction (CSP) and complimentary greedy search-based prediction (CGSP) are given when 20, 30, or all SNPs are chosen as informative SNPs.
ROC Curve Comparison of 5 prediction methods on (Barkash et. al,2006 ) data on all SNPs. Area under the CSP’s curve is 0.87 vs 0.52 under the SVM’s curve.
Thank you! Dr. Alexander Zelikovsky Ph.D students: Jingwu He Weidong Mao Nisar Hundewale Dumitru Brinza Stefan Gremalschi Kelly Westbrooks
Quality Measures of Prediction (confusion table) • Sensitivity: The ability to correctly detect disease. sensitivity = TP/(TP+FN) • Specificity: The ability to avoid calling normal as disease. specificity = TN/(FP+TN) • Accuracy = (TP +TN)/(TP+FP+FN+TN) • Risk Rate: Measurements for risk factors.
Cross-validation Method • Leave-one-out test: The disease status of each genotype in the data set is predicted while the rest of the data is regarded as the training set. Predicted Disease Status Real Disease Status Genotype -1 -1 0101201020102210 -1 -1 0220110210120021 Accuracy = 80% -1 1 0200120012221110 1 1 0020011002212101 1 1 0020011002212101 • Leave-many-out test: Repeat randomly picking 2/3 of the population as training set and predict the other 1/3.
Algorithms Evaluation • P-value: probability of null hypotheses. • Null hypotheses: The observed prediction accuracy is obtained by chance. • To reject the null hypotheses, p-value < 0.05 • Unadjusted p-value: Probability of case/control distribution in a bucket, computed by binomial distribution • Multiple-testing adjusted p-value : randomization • Randomly permute the disease status of the population to generate 1000 instances. • Apply prediction methods on each instance to get prediction accuracy. • Compute the probability of instances that have a higher prediction accuracy than the observed accuracy.
Most disease-associated & disease-resistant MSC Comparison of three methods for searching the disease-associated and disease- resistant multi-SNPs combinations with the largest PPV. The starred values refer to results of the runtime-constrained exhaustive search