Download

1 / 56
560 likes | 704 Views
A Prototype Finite Difference Model. Part 1: Stommel Model – definition and serial implementation. 2D solution to a model problem for Ocean Circulation

E N D
Part 1: Stommel Model – definition and serial implementation • 2D solution to a model problem for Ocean Circulation • Serves as a prototype finite difference model. Illustrates many aspects of a typical code taking advantage of distributed memory computers using MPI.
The Stommel Problem • Wind-driven circulation in a homogeneous rectangular ocean under the influence of surface winds, linearized bottom friction, flat bottom and Coriolis force. • Solution: intense crowding of streamlines towards the western boundary caused by the variation of the Coriolis parameter with latitude
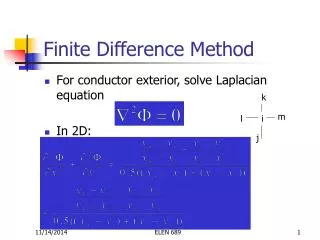
Governing Equations 2 2 ¶ y ¶ y ¶ y ( ) f b g + + = 2 2 x ¶ x y ¶ ¶ y p f s i n ( ) = - a 2L y L L 2 0 0 0 K m = = x y ( 6 ) - 3 1 0 g = * ( 1 1 ) - 2 . 2 5 1 0 b = * ( 9 ) - 1 0 a =
Domain Discretization D e f i n e a g r i d c o n s i s t i n g o f p o i n t s ( x , y ) g i v e n b y i j x i x , i 0 , 1 , . . . . . . . , n x + 1 = D = i y j y , j 0 , 1 , . . . . . . . , n y + 1 = D = j x L ( n x 1 ) D = + x y L ( n y 1 ) D = + y
Domain Discretization (cont’d) Seek to find an approximate solution ( x , y ) a t p o i n t s ( x , y ) : y i i j j ( x , y ) y » y i , j i j
Centered Finite Difference Scheme for the Derivative Operators
Five-point Stencil Approximationfor the Discrete Stommel Model i, j+1 interior grid points: i=1,nx; j=1,ny boundary points: (i,0) & (i,ny+1) ; i=1,nx (0,j) & (nx+1,j) ; j=1,ny i, j i-1, j i+1, j i, j-1
Jacobi Iteration Start with an initial guess for do i = 1, nx; j = 1, ny end do Copy Repeat the process
Implementation Overview /work/Training/examples/stommel/ • Code written in Fortran 90 as well as in C • Uses many features of F90 • Modules instead of commons • Free format • Module with kind precision facility • Interfaces • Allocatable arrays • Array syntax
Free Format • Statements can begin in any column • ! Starts a comment • To continue a line use a “&” on the line to be continued
Modules instead of commons • Modules have a name and can be used in place of named commons • Modules are defined outside of other subroutines • To “include” the variables from a module in a routine you “use” it • The main routine stommel and subroutine jacobi share the variables in module “constants” module constants real dx,dy,a1,a2,a3,a4,a5,a6 end module … program stommel use constants … end program subroutine jacobi use constants … end subroutine jacobi
Kind precision facility Instead of declaring variables real*8 x,y We use real(b8) x,y Where b8 is a constant defined within a module module numz integer,parameter::b8=selected_real_kind(14) end module program stommel use numz real(b8) x,y x=1.0_b8 ...
Why use kind precision facility? Legality Portability Reproducibility Modifiability real*8 x,y is not legal syntax in Fortran 90 Declaring variables “double precision” will give us 16 byte reals on some machines integer,parameter::b8=selected_real_kind(14) real(b8) x,y x=1.0_b8uprecision
Allocatable arrays • We can declare arrays to be allocatable • Allows dynamic memory allocation • Define the size of arrays at run time real(b8),allocatable::psi(:,:) ! our calculation grid real(b8),allocatable::new_psi(:,:) ! temp storage for the grid ! allocate the grid to size nx * ny plus the boundary cells allocate(psi(0:nx+1,0:ny+1)) allocate(new_psi(0:nx+1,0:ny+1))
Interfaces • Similar to C prototypes • Can be part of the routines or put in a module • Provides information to the compiler for optimization • Allows type checking module face interface bc subroutine bc (psi,i1,i2,j1,j2) use numz real(b8),dimension(i1:i2,j1:j2):: psi integer,intent(in):: i1,i2,j1,j2 end subroutine end interface end module program stommel use face ...
Array Syntax Allows assignments of arrays without do loops ! allocate the grid to size nx * ny plus the boundary cells allocate(psi(0:nx+1,0:ny+1)) allocate(new_psi(0:nx+1,0:ny+1)) ! set initial guess for the value of the grid psi=1.0_b8 ! copy from temp to main grid psi(i1:i2,j1:j2)=new_psi(i1:i2,j1:j2)
Program Outline (Fortran) (1) • Module NUMZ - defines the basic real type as 8 bytes • Module INPUT - contains the inputs • nx,ny (Number of cells in the grid) • lx,ly (Physical size of the grid) • alpha,beta,gamma (Input calculation constants) • steps (Number of Jacobi iterations) • Module Constants - contains the invariants of the calculation
Program Outline (2) • Module face - contains the interfaces for the subroutines • bc - boundary conditions • do_jacobi - Jacobi iterations • force - right hand side of the differential equation • Write_grid - writes the grid
Program Outline (3) • Main Program • Get the input • Allocate the grid to size nx * ny plus the boundary cells • Calculate the constants • Set initial guess for the value of the grid • Set boundary conditions • Do the Jacobi iterations • Write out the final grid
C version considerations • To simulate the F90 numerical precision facility we: • #define FLT double • And use FLT as our real data type throughout the rest of the program • We desire flexibility in defining our arrays and matrices • Arbitrary starting indices • Contiguous blocks of memory for 2d arrays • Use routines based on Numerical Recipes in C
Vector allocation routine FLT *vector(INT nl, INT nh) { /* creates a vector with bounds vector[nl:nh]] */ FLT *v; /* allocate the space */ v=(FLT *)malloc((unsigned) (nh-nl+1)*sizeof(FLT)); if (!v) { printf("allocation failure in vector()\n"); exit(1); } /* return a value offset by nl */ return v-nl; }
Matrix allocation routine FLT **matrix(INT nrl,INT nrh,INT ncl,INT nch) /* creates a matrix with bounds matrix[nrl:nrh][ncl:nch] */ /* modified from the book version to return contiguous space */ { INT i; FLT **m; /* allocate an array of pointers */ m=(FLT **) malloc((unsigned) (nrh-nrl+1)*sizeof(FLT*)); if (!m){ printf("allocation failure 1 in matrix()\n"); exit(1);} m -= nrl; /* offset the array of pointers by nrl */ for(i=nrl;i<=nrh;i++) { if(i == nrl){ /* allocate a contiguous block of memroy*/ m[i]=(FLT *) malloc((unsigned) (nrh-nrl+1)*(nch-ncl+1)*sizeof(FLT)); if (!m[i]){ printf("allocation failure 2 in matrix()\n");exit(1); } m[i] -= ncl; /* first pointer points to beginning of the block */ } else { m[i]=m[i-1]+(nch-ncl+1); /* rest of pointers are offset by stride */ } } return m; }
Compiling and running (serial) Compile: xlf90 -O4 stf_00.f -o stf xlc -O4 stc_00.c -o stc Run: poe stf < st.in -nodes 1 -tasks_per_node 1 -rmpool 1 Exercise: measure run times of Fortran and C versions, both optimized (-O4) and unoptimized. Draw conclusions.
Part 2: MPI Version of the Stommel Model code with 1D Decomposition • Choose one of the two dimensions andsubdivide the domain to allow the distribution of the work across a group of distributed memory processors. • We will focus on the principles and techniques usedtodo the MPI work in the model. • Fortran version is used in the presentation.
Introduce the MPI environment • Need to include “mpif.h” to define MPI constants • Need to define our own constants: npes - how many processors are running myid - which processor I am ierr - error code returned by most calls pi_master - the id for the master node Add the following lines: include "mpif.h" integer npes,myid,ierr integer, parameter::mpi_master=0
MPI environment (cont’d) Next need to initialize MPI - add the following to the beginning of your program: call MPI_INIT( ierr ) call MPI_COMM_SIZE(MPI_COMM_WORLD,npes, ierr) call MPI_COMM_RANK(MPI_COMM_WORLD, myid, ierr) write(*,*)’from ‘, myid,’npes=‘,npes At the end of the program add: call MPI_Finalize(ierr) stop
Try it! Compile mpxlf90 –O3 stf_00mod.f -o st_test mpcc –O3 stf_00mod.c -o st_test Run poe st_test < st.in –nodes 1 –tasks_per_node 2 –rmpool 1 Try running again. Do you get the same output? What does running on 2 CPUs really do?
Modify data input We read the data on processor 0 and send to the others if(myid .eq. mpi_master)then read(*,*)nx,ny read(*,*)lx,ly read(*,*)alpha,beta,gamma read(*,*)steps endif We use MPI_BCAST to send the data to the other processors - 8 calls -- one for each variable.
Domain Decomposition (1-D)Physical domain is sliced into sets of columns so that computation in each set of columns will be handled by different processors (In C subdivide in rows) . Why do columns and not rows? Parallel version each processor gets half the cells Serial version all cells on one processor CPU #1 CPU #2
Domain Decomposition (1-D) Fortran 90 allows us to set arbitrary bounds on arrays. Use this feature to break the original grid psi(1:nx,1:ny) into npes subsections of size ny/npes. For 2 CPUs we get the following: CPU#1: psi(1:nx,1:ny/2) CPU#2: psi(1:nx,ny/2+1,ny) • No two processors calculate psi for the same (i,j) • For every processor, psi(i,j) corresponds to psi(i,j) in the serial program. What other ways of doing decomposition can you think of?
Domain Decomposition (1-D) What are ghost cells? To calculate the value for psi(4,4) CPU #1 requires the value from psi(4,3),psi(5,4),psi(3,4),psi(4,5) CPU#1 needs to get (receive) the value of psi(4,5) from CPU#2. CPU#1 will hold the value of psi(4,5) in one of it’s ghost cells. Ghost cells for CPU#1 are created by adding an extra column to the psi array: instead of psi(1:8,1:4) have psi(1:8,1,5) where elements psi(*,5) are ghost cells used to store psi values from CPU#2 CPU #1 CPU #2
Domain Decomposition (1-D) With ghost cells our decomposition becomes... Parallel version each processor gets half the cells plus ghost cells Serial version all cells on one processor CPU #1 CPU #2 ghost cells
Domain Decomposition (1-D) Source code for setting up the distributed grid with ghost cells ! Set the grid indices for each CPU i1=1 i2=nx dj=real(ny)/real(npes) j1=nint(1.0+myid*dj) j2=nint(1.0+(myid+1)*dj)-1 write(*,101)myid,i1,i2,j1,j2 101 format("myid= ",i3,3x,& " (",i3," <= i <= ",i3,") , ", & " (",i3," <= j <= ",i3,")") ! allocate the grid to size i1:i2,j1:j2 but add additional ! cells for boundary values and ghost cells allocate(psi(i1-1:i2+1,j1-1:j2+1)) Since 1-D decomposition only the y-indices change Added to display each CPU’s indices
Ghost cell update Each trip through our main loop we call do_transfer to update the ghost cells. Our main loop becomes... do i=1,steps call do_jacobi(psi,new_psi,mydiff,i1,i2,j1,j2,& for,nx,ny,a1,a2,a3,a4,a5) !update ghost cell values call do_transfer(psi,i1,i2,j1,j2,myid,npes) write(*,*)i,diff enddo
Odd step2 Odd step1 How do we update ghost cells? • Processors send values to and receive from their neighbors. • Need to exchange with left and right neighbors, except processors on far left and right only transfer in 1 direction • Trick 1: to avoid deadlock where all processor try to send, need to split up calls to send and receive: • Even numbered processors: Odd numbered processors: • send to left receive from right • receive from left send to right • receive from right send to left • send to right receive from left Even step1 Odd step1 Even step2 Odd step2 Even step1 Even step2 Trick2: To handle the end CPUs send to MPI_PROC_NULL instead of a real CPU
How do we update ghost cells? ! How many cells are we sending num_x=i2-i1+3 ! Where are we sending them myleft=myid-1 !processor id of CPU on the left myright=myid+1 !processor id of CPU on the right if(myleft .le. -1)myleft=MPI_PROC_NULL if(myright .ge. npes)myright=MPI_PROC_NULL
How do we update ghost cells?For even-numbered processors... if(even(myid))then ! send to left call MPI_SEND(psi(:,j1),num_x,MPI_DOUBLE_PRECISION,& myleft,100,MPI_COMM_WORLD,ierr) ! rec from left call MPI_RECV(psi(:,j1-1),num_x,MPI_DOUBLE_PRECISION,& myleft,100,MPI_COMM_WORLD,status,ierr) ! rec from right call MPI_RECV(psi(:,j2+1),num_x,MPI_DOUBLE_PRECISION,& myright,100,MPI_COMM_WORLD,status,ierr) ! send to right call MPI_SEND(psi(:,j2), num_x,MPI_DOUBLE_PRECISION,& myright,100,MPI_COMM_WORLD,ierr) else
How do we update ghost cells?For odd-numbered processors... Else ! we are on an odd column processor ! rec from right call MPI_RECV(psi(:,j2+1),num_x,MPI_DOUBLE_PRECISION, & myright,100,MPI_COMM_WORLD,status,ierr) ! send to right call MPI_SEND(psi(:,j2), num_x,MPI_DOUBLE_PRECISION, & myright,100,MPI_COMM_WORLD,ierr) ! send to left call MPI_SEND(psi(:,j1), num_x,MPI_DOUBLE_PRECISION, & myleft,100,MPI_COMM_WORLD,ierr) ! rec from left call MPI_RECV(psi(:,j1-1),num_x,MPI_DOUBLE_PRECISION, & myleft,100,MPI_COMM_WORLD,status,ierr) endif
How do we update ghost cells?It’s a 4-stage operation.Example with 4 CPUs:
Other modificationsForce and do_jacobi are not modifiedModify the boundary condition routine so that it only sets values for true boundaries and ignore ghost cells subroutine bc: ! check which CPUs have the edges and assign them the ! correct boundary values if(i1 .eq. 1) psi(i1-1,:)=0.0 !top edge if(i2 .eq. nx) psi(i2+1,:)=0.0 !bottom edge if(j1 .eq. 1) psi(:,j1-1)=0.0 !left edge if(j2 .eq. ny) psi(:,j2+1)=0.0 !right edge Only add if-then statements
Other Modifications : Residual • In our serial program, the routine do_jacobi calculates a residual for each iteration • The residual is the sum of changes to the grid for a jacobi iteration • Now the calculation is spread across all processors • To get the global residual, we can use the MPI_Reduce function call MPI_REDUCE(mydiff,diff,1,MPI_DOUBLE_PRECISION, & MPI_SUM,mpi_master,MPI_COMM_WORLD,ierr) if(myid .eq. mpi_master)write(*,*)i,diff (Since mpi_master now has value of diff, only that CPU writes value)
Our main loop is now…Call the do_jacobi subroutineUpdate the ghost cellsCalculate the global residual do i=1,steps call do_jacobi(psi,new_psi,mydiff,i1,i2,j1,j2) call do_transfer(psi,i1,i2,j1,j2) call MPI_REDUCE(mydiff,diff,1,MPI_DOUBLE_PRECISION, & MPI_SUM,mpi_master,MPI_COMM_WORLD,ierr) if(myid .eq. mpi_master)write(*,*)i,diff enddo
Final change • We change the write_grid subroutine so that each node writes its part of the grid to a different file. • Function unique returns a file name based on a input string and the node number We change the open statement in write_grid to: open(18,file=unique("out1d_"),recl=max(80,15*((jend-jstart)+3)+2))
Unique Unique is the function: function unique(name,myid) implicit none character (len=*) name character (len=20) unique character (len=80) temp integer myid if(myid .gt. 99)then write(temp,"(a,i3)")trim(name),myid else if(myid .gt. 9)then write(temp,"(a,'0',i2)")trim(name),myid else write(temp,"(a,'00',i1)")trim(name),myid endif endif unique=temp return end function unique
Suggested exercises • Study, compile, and run the program stf_01.f and/or stc_01.c on various numbers of processors (2--8). Note timing. • Do decompositionin rows if using Fortran, columns if using C). • Do decomposition so that psi starts at i=1,j=1 for each processor. • Do periodic boundary conditions. • Modify the write_grid routine to output the whole grid from CPU#0
Part 3: 2-D decomposition • The program is almost identical • We now have our grid distributed in a block fashion across the processors instead of striped • We can have ghost cells on 1, 2, 3 or 4 sides of the grid held on a particular processor
2-D Decomposition Example 2-D Decomposition on 50 x 50 grid on 4 processors: CPU #1 CPU #0 Grid on each processor is allocated to: pid= 0 ( 0 <= i <= 26) , ( 0 <= j <= 26) pid= 1 ( 0 <= i <= 26) , ( 25 <= j <= 51) pid= 2 ( 25 <= i <= 51) , ( 0 <= j <= 26) pid= 3 ( 25 <= i <= 51) , ( 25 <= j <= 51) CPU #3 CPU #2 But each processor calculates only for: Where interior grid size is 25,25 pid= 0 ( 1 <= i <= 25) , ( 1 <= j <= 25) pid= 1 ( 1 <= i <= 25) , ( 26 <= j <= 50) pid= 2 (26 <= i <= 50) , ( 1 <= j <= 25) pid= 3 (26 <= i <= 50) , ( 26 <= j <= 50) Extra cells are ghost cells
Only three changes need to be made to our program 1. Given an arbitrary number of processors, find a good topology (number of rows and columns of processors) 2. Make new communicators to allow for easy exchange of ghost cells • Set up communicators so that every processor in the same row is in a given communicator • Set up communicators so that every processor in the same column is in a given communicator 3. Add the up/down communication to do_transfer