Download

1 / 31
370 likes | 857 Views
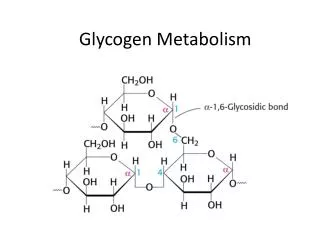
Glycogen. Glycogen. Liver ~ 4 % 72 g Muscle ~ 1 % 245 g Extracellular glucose 0,1 % 10 g Adult - man 70 kg His liver 1,8 kg His muscle mass 35 kg Blood volume 10 l. Glycogen. Muscle: Glucose substrate for muscle glycolysis Liver: Glucose store, Export of hexose Blood sugar

E N D
Glycogen • Liver • ~ 4 % 72 g • Muscle • ~ 1 % 245 g • Extracellular glucose • 0,1 % 10 g Adult - man 70 kg His liver 1,8 kg His muscle mass 35 kg Blood volume 10 l
Glycogen • Muscle: • Glucose substrate for muscle glycolysis • Liver: • Glucose store, • Export of hexose • Blood sugar • Glycogenosis • Deff. Of enzymes • Muscle weakness ¢N
As a meal containing carbohydrates is eaten and digested, • blood glucose levels rise • pancreas secretes insulin. • Glucose from the portal vein enters the liver cells (hepatocytes). • Insulin acts on the hepatocytes to stimulate the action of several enzymes, including glycogen synthase. • Glucose molecules are added to the chains of glycogen as long as both insulin and glucose remain plentiful. In this postprandial or "fed" state, the liver takes in more glucose from the blood than it releases. • .
After a meal has been digested and glucose levels begin to fall, insulin secretion is reduced, and glycogen synthesis stops. • About four hours after glycogen begins to be broken down and converted again to glucose.
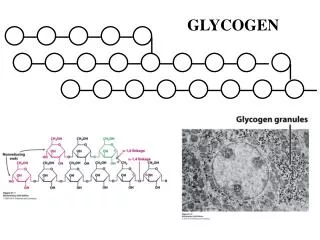
Glycogen synthesis • Muscle • Hexokinase • Liver • Glucokinase • Phosphoglucomutase • Glucose-1,6-P cofactor • Glycogen • Primer: glycogenin • 37 kDa glycosilated Tyr
Synthesis of glycogen glikogenin
Glycogen P P P Glygenolysis Glycogenesis Glucose-1-P
Effect of epinephrin X Stimulus R cAMP 40X ATP Cent. Nervous syst. Inakt. Protein Kinase A Aktive Protein Kinase A 10X Adrenal cortex Phosphorylase Kinaseb Phosphorylase Kinase a 100X Glykogen phosphorylaseb Glykogen phosphorylasea 1000X Epinephrin X Glykogen Glukose 1 P 10000X
GDP b a g ATP GTP cAMP
4 cAMP R R R R C C C C
Protein Kinase Reactions • Serine-Threonine Kinase / cAMP depentent Protein Kinase
Tein, koffein cAMP AMP PKA PKA Phosphorylase-kinase b Phosphorylase-kinase a Ca2+ Phosphorylase a Phosphorylase b AMP
Glycogen P P P Glycogenolysis Glycogenesis Glucose-1-P
Glycogen synthase • Glycogen synthasea –dephosphorylated • Glycogen synthaseb –phosphorylated • 4 identical subunit • 7 Ser-OH residues/subunit
Glycogen synthase • 6 different protein kinases • Phophorylase kinase (Ca2+/Calmodulin dep) • Ca2+/Calmodulin dep. • GSK-3: • GSK-4 • GSK-5 • Glucose-6-P: allosteric activator of Glycogene-synthase kinaseb • Insulin/muscle: • dephosphorylation / activation of Glycogen-synthaseb
Von Gierke’s Disease Glucose 6-phosphatase: liver and kidney Here is another liver with a pale, bulging surface. This time the liver is filled with glycogen in von Gierke's disease, the glycogen storage disease of children. Severly enlarged liver, severe hypoglycemia, lactic acidosis, ketosis, hyperuricemia, hyperlipemia
POMPE'S DISEASE When mannose 6-phosphate tags are added to acid maltase enzyme molecules, the molecules stick to receptors (docking sites) on the muscle cells and are carried deeper inside the cells, where they're needed. Without mannose 6-phosphate tags, acid maltase (1,4-D-Glucosidase) enzyme molecules can't enter muscle cells from the bloodstream. ENZYME TREATMENT BENEFITS BABIES WITH POMPE'S DISEASE Babies with a metabolic muscle disorder known as Pompe's disease, or acid maltase deficiency, usually don't survive infancy because they lack a vital enzyme that normally breaks down glycogen in the heart and skeletal muscle cells. Cardiac failure in infancy; liver, heart, muscle
Forbe’s disease, Type III glycogenosis • also called Cori's disease , or glycogenosis type III rare hereditary disease in which the the metabolic breakdown of glycogen to the simple sugar glucose is incomplete, allowing intermediate compounds to accumulate in the cells of the liver. Affected persons lack the enzyme amylo-1,6-glucosidase, one of several enzymes involved in glycogen breakdown. Children with the disease have enlarged livers (which usually…
Andersen’s disease • also called Glycogenosis Type Iv, extremely rare hereditary metabolic disorder produced by absence of the enzyme amylo-1:4,1:6-transglucosidase (branching enzyme), which is an essential mediator of the synthesis of glycogen. An abnormal form of glycogen, amylopectin, is produced and accumulates in body tissues, particularly in the liver and heart. Affected children appear normal at birth but fail to thrive and later lose… Liver cirrhosis, death usually before 24 months
McArdle's Disease Phosphorylase stain: Absent Muscle fibers stain yellowMyophosphorylase deficiency: McArdle's disease Phosphorylase stain: Normal more darkly than type I