Download
1 / 13
140 likes | 165 Views
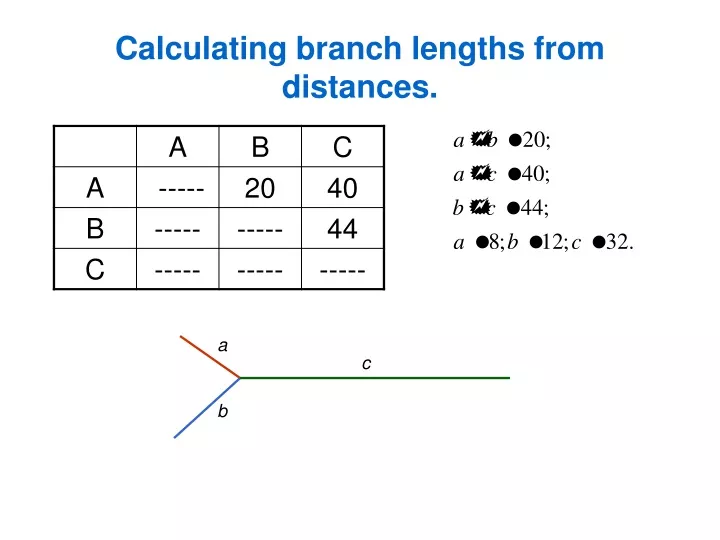
Calculating branch lengths from distances. a. c. b. 1.1 Distance methods: Neighbor-joining method. NJ is based on minimum evolution principle (sum of branch length should be minimized).

E N D
1.1 Distance methods: Neighbor-joining method. NJ is based on minimum evolution principle (sum of branch length should be minimized). Given the distance matrix between all sequences, NJ joins sequences in a tree so that to give the estimate of branch lengths. • Starts with the star tree, calculates the sum of branch lengths. C B b c D a d e A E
1.2 Neighbor-joining method. 2. Combine two sequences in a pair, modify the tree. Recalculate the sum of branch lengths, S for each possible pair, choose the lowest S. C B c b d D a e A E 3. Treat cluster CDE as one sequence “X”, calculate average distances between “A” and “X”, “B” and “X”, calculate “a” and “b”. 4.Treat AB as a single sequence, recalculate the distance matrix. 5. Repeat the cycle and calculate the next pair of branch lengths.
2.1 Maximum parsimony: definition of informative sites. Maximum parsimony tree – tree, that requires the smallest number of evolutionary changes to explain the differences between external nodes. Site, which favors some trees over the others. 1 2 3 4 5 6 7 A A G A C T G A G C C C T G A G A T T T C A G A G T T C * * Site is informative if there are at least two different kinds of letters at the site, each of which is represented in at least two of the sequences.
2.2 Maximum parsimony. Site 3 1.G 3.A 1.G 2.C 2.C 1.G G A A A A A 2.C 4.A 3.A 4.A 4.A 3.A Tree 1. Tree 2. Tree 3. Site 3 is not informative, all trees are realized by the same number of substitutions. Advantage: deals with characters, don’t need to compute distance matrices. Disadvantage: - multiple substitutions are not considered - branch lengths are difficult to calculate - slow
2.3 Maximum parsimony method. • Identify all informative sites in the alignment. 2. Calculate the minimum number of substitutions at each informative site. 3. Sum number of changes over all informative sites for each tree. 4. Choose tree with the smallest number of changes.
Maximum likelihood methods. • Similarity with maximum parsimony: - for each column of the alignment all possible trees are calculated - trees with the least number of substitutions are more likely • Advantage of maximum likelihood over maximum parsimony: - takes into account different rates of substitution between different amino acids and/or different sites - applicable to more diverse sequences
Molecular clock. • First observation: rates of amino acid substitutions in hemoglobin and cytochrome c are ~ the same among different mammalian lineages. • Molecular clock hypothesis: rate of evolution is ~ constant over time in different lineages; proteins evolve at constant rates. • This hypothesis is used in estimating divergence times and reconstruction of phylogenetic trees.
Estimation of species divergence time. Assumption: rate constancy, molecular clock. Find T1 if T2 is known. T1 T2 A B C
Fixation of mutations. Not all mutations are spread through population. Fixation – when a mutation is incorporated into a genome of species. Fixation rate will depend on the size of population (N), fitness (s) and mutation rate (μ):
Neutral theory of evolution. • Kimura in 1968: majority of molecular changes in evolution are due to the random fixation of neutral mutations (do not effect the fitness of organism. • As a consequence the random genetic drift occurs. • Value of selective advantage of mutation should be stronger than effect of random drift.
Classwork: maximum marsimony. • Search the NCBI Conserved Domain Database for pfam00127. • Construct maximum parsimony tree using MEGA3. • Analyze this tree and compare it with the phylogenetic tree from the research paper.