Download

1 / 18
200 likes | 1.21k Views
HEMOGLOBİN TAYİNİ, HEMOGLOBİN YAPISI, HEMOGLOBİNDE FARKLILIKLAR. Doç.Dr. Mustafa ALTINIŞIK ADÜTF Biyokimya AD 2007. Hemoglobin tayini. Hemoglobin tayini çeşitli metodlarla yapılabilmektedir: -Sahli metodu -Siyanmethemoglobin metodu -Oksihemoglobin metodu -Elektronik sayıcılarla ölçüm .

E N D
HEMOGLOBİN TAYİNİ, HEMOGLOBİN YAPISI, HEMOGLOBİNDE FARKLILIKLAR Doç.Dr. Mustafa ALTINIŞIK ADÜTF Biyokimya AD 2007
Hemoglobin tayini Hemoglobin tayini çeşitli metodlarla yapılabilmektedir: -Sahli metodu -Siyanmethemoglobin metodu -Oksihemoglobin metodu -Elektronik sayıcılarla ölçüm
Sahli Metodu ile Hemoglobin Tayini Hemoglobinin hidroklorik asit ile asit hematine dönüşmesi ve koyu kahverengi-sarı renk alması prensibine dayanır. Sahli hemoglobinometresi, Sahli hemoglobin pipeti (0.02 ml), Hidroklorik asit (%1’lik), distile su, lanset, alkol, pamuk gerekli araç ve gereçlerdir.
-Sahli hemoglobinometresindeki üzeri dereceli tüpe 5 damla %1’lik hidroklorik asit konur. -Parmak ucu steril şartlarda lanset ile delinir. Buradan veya antikoagulanlı kandan Sahli pipetine 20 mikron (0.02 ml) işaretine kadar kan çekilir, pipetin etrafı pamukla temizlenir. -Pipetteki kan tüpteki hidroklorik asit içine boşaltılır. Pipet birkaç kere çekip boşaltılarak kanın tümünün asit ile karışması sağlanır.
-Hemoglobinin hidroklorik asit ile asit hematine dönüşmesi için birkaç dakika beklenir. -Tüpteki karışıma damla damla distile su eklenir ve karıştırılır. -Sulandırma işlemine, tüpteki karışımın rengi hemoglobinometrenin her iki yanındaki standart rengi alıncaya kadar devam edilir. -Hemoglobinometre tüpündeki sıvının seviyesine uyan hemoglobin, % veya gram olarak okunur. %100 hemoglobin, 16 g/dl hemoglobin demektir.
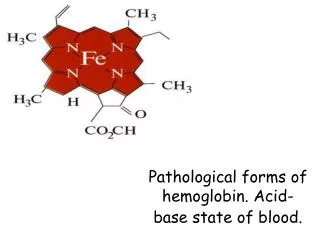
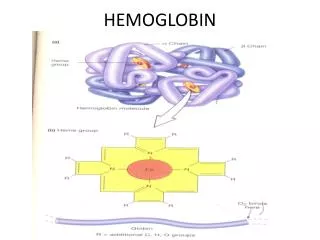
Hemoglobin yapısı Hemoglobin molekülü 4 hem ve 1 globin içerir.
Hemoglobindeki 4 hemin her biri bir protoporfirin III ve bir Fe2+ içerir.
Hemoglobinin protein komponenti olan globin, glisince fakir bazik amino asitlerce zengin bir proteindir; tetrahedron şekilde düzenlenmiş 4 polipeptit zincirden yapılmıştır.
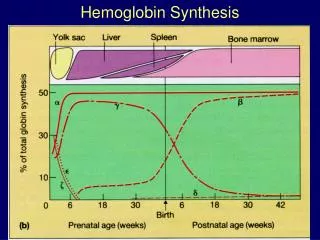
Çeşitli hemoglobin tiplerinde bulunabilen polipeptit zincirleri -zincir, -zincir, -zincir, -zincir olmak üzere dört tiptir. Fizyolojik hemoglobinler, erişkin bir şahsın kanındaki eritrositlerde bulunan HbA1, HbA2, HbF hemoglobinleridirler.
HbA1: Globininde 2 ve 2polipeptit zinciri içeren fizyolojik hemoglobindir. HbA1,erişkin bir şahsın eritrositlerinde bulunan hemoglobinin %97-98’ini oluşturur. HbA2: Globininde 2 ve 2polipeptit zinciri içeren fizyolojik hemoglobindir.HbA2, erişkin sağlıklı bir şahsın eritrositlerinde bulunan hemoglobinin %0,5-2,5’ini oluşturur. HbF: Globininde 2 ve 2polipeptit zinciri içeren, primitif hemoglobin (HbP) diye de bilinen fizyolojik hemoglobindir. HbF, erişkin sağlıklı bir şahsın eritrositlerinde bulunan hemoglobinin %1’inden azını oluşturur.
Çeşitli patolojiler anormal hemoglobinlerin ortaya çıkmasına neden olur: 1) Hemoglobinin polipeptit zincirine bir veya daha fazla amino asit eklenebilir, zincirden amino asit çıkabilir veya zincirdeki amino asitler yer değiştirebilir. 2) Globin zinciri üretiminde defekt olabilir; belli bir globin zinciri türü üretilmez. 3) Tip 1 ve tip 2’nin kombinasyonu olabilir. 4) Herediter persistant fetal hemoglobinemi de tanımlanmıştır; asemptomatiktir.
Anormal hemoglobinlerin pek çok çeşidi vardır ki bunlar, keşfedildiği yer, ilk keşfedilen ailedeki isimler ya da yer değişimi olan amino asidin adı temel alınarak isimlendirilmektedirler: Hb S:HbA1’in -zincirlerindeki 6. amino asit glutamik asit değil de valin olan hemoglobindir. Hb S, orak hücreli anemi (Hb S hastalığı) olarak tanımlanan hemoglobinopatinin ortaya çıkmasına neden olur.
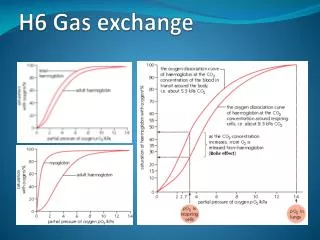
Orak hücreli anemi ( Hb S hastalığı), otozomal resesif kalıtılır. Homozigotlarda hemolitik anemi ortaya çıkar. Heterozigot taşıyıcılarda asemptomatiktir; ancak pO2 25 mmHg’nın altına düştüğünde oraklaşmış hücreler dalakta toplanırlar, dolaşımı bozarlar, ağrılı damar tıkanıklıklarına neden olurlar.
Hb C: HbA1’in -zincirlerindeki 6. amino asit glutamik asit değil de lizin olan hemoglobindir. Hb C, hemoglobin C hastalığı olarak bilinen hemoglobinopatinin ortaya çıkmasına neden olur. Bir kısmı globinde orak hücre mutasyonu gösterirken bir kısmı Hb C özelliğinde olan bir hemoglobinopati, hemoglobin SC hastalığı olarak tanımlamaktadır.
Hb H ve Hb Bart:Hb H, globini -zinciri içermeyen, 4-zinciri içeren hemoglobindir. Hb Bart ise globini -zinciri içermeyen, 4-zinciri içeren hemoglobindir. Hb H ve Hb Bart, -talasemili hastaların kanında saptanır. Talassemiler, hipokrom mikrositer anemilerdir. Talassemiler,herediter hemolitik hastalıklardır; insanlarda tek gen defektinin görüldüğü en sık rastlanan hastalık grubunu oluştururlar. Talassemilerde, normalde uyum içinde sentezlenen ve zincirlerinden biri sentezlenmez veya azalmış olarak sentezlenir.
-talassemilerde globin zincirlerinin sentezi ya hiç olmaz ya da azalmıştır. Her birey, globin geni için, her bir 16.kromozomda 2 adet olmak üzere dört kopya içerir. Bir gen defektli ise, “silent carrier” terimi kullanılır; belirti yoktur. İki globin geninde defekt varsa “ talassemi trait” söz konusudur. Üç globin geninde defekt varsa “hemoglobin H hastalığı” söz konusudur; orta dereceden ciddiye doğru değişen hemolitik anemi tablosu sergilenir. globin genlerinin dördü de defektli ise fetal ölümle sonuçlanan “hidrops fetalis” ortaya çıkar;çünkü globin zincirleri, HbF oluşumu için de gereklidir.
-talassemilerde globin zincirlerinin sentezi azalır veya olmaz; globin zincir sentezi normaldir. Her birey, globin geni için, her bir 11.kromozomda 1 adet olmak üzere iki kopya içerir. Tek gen defekti olduğunda “-talassemi minör” söz konusudur. Her iki globin geninde defekt olduğunda “-talassemi majör” söz konusudur. -talassemi majörde hastalar, yaşamın bir veya ikinci yılında ciddi anemi tablosu sergilerler; bu hastalar için düzenli kan transfüzyonu gerekir; kronik kan tranfüzyonu nedeniyle hastalarda demir yüklenmesine bağlı “hemosiderozis” tablosu gelişir.
Talassemi (Akdeniz Anemisi), Mendelian kurallarına göre otozomal resesif kalıtım özelliğine sahiptir. Burada bir kalıtımsal taşıyıcılık kavramı söz konusudur.Yani toplumda hiçbir hastalık bulgusu taşımayan, tamamen normal bir yaşam süresi ve yaşam kalitesine sahip bazı bireyler bu genetik defekti çekinik bir karakter olarak taşımaktadırlar. Taşıyıcı birey normal bir birey ile evlenirse doğacak çocukları, normal veya taşıyıcı olacaktır. Yani görünür bir sorun olmayacaktır. Ancak bu taşıyıcı bireyler yine kendileri gibi taşıyıcı bir bireyle evlenirlerse, işte o zaman her doğacak çocukları için 1/4 yani %25 hasta doğma olasılığı vardır.