Download

1 / 38
400 likes | 965 Views
1. Protein Docking. Park, Jong Hwa MRC-DUNN Hills Road Cambridge CB2 2XY England. Bioinformatics in Biosophy. Next :. 02/06/2001. Defining the problem of docking (from Amit Singh’s talk slides). Docking is one of the hardest problems.

E N D
1 Protein Docking Park, Jong Hwa MRC-DUNN Hills Road Cambridge CB2 2XY England Bioinformatics in Biosophy Next: 02/06/2001
Defining the problem of docking (from Amit Singh’s talk slides)
Docking is one of the hardest problems Protein Docking is an extension of protein folding. Computationally very expensive. Automatic prediction of docking is an important step toward rational drug design. Docking is different from interaction. It is a high resolution analysis and prediction for protein function. (It is a part of Interaction research)
The difference between Interaction and Docking Interaction is a more general and higher level concept. It includes gene-gene, protein-protein, protein ligand interactions in both Direct and Indirect structural/non-structural interaction. Docking is PHYSICAL and involves Structural interaction only.
Types of docking Protein Protein Docking Protein Ligand Docking (Ligand Ligand Docking)
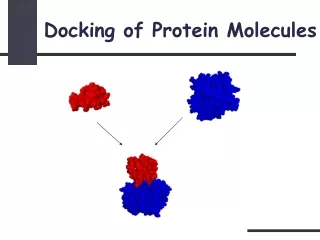
Protein Protein Docking Surface vs Surface interaction of two or more proteins. 1. Homo Interaction 2. Hetero Docking of protein structures.
Techniques: Geometric hashing The algorithm is for object recognition. Given a scene containing objects and a database of models, this algorithm analyzes the objects to detect whether any of the models of the database are present in the scene. The models returned by the algorithm and the corresponding objects in the scene can then be analyzed more carefully to validate the matches. This algorithm is originally developed in the robotics-vision communities, but it has found applications in other domains, such as medical image registration, detection of defects in the boundary of objects in CAD models, and ligand-protein or protein-protein docking in structural biology. This summary will attempt to give an overview of the algorithm, along with how it is applied in the domain of structural biology.
FlexX FlexX is a computer program for predicting protein-ligand interactions. For a protein with known three-dimensional structure and a small ligand molecule, FlexX predicts the geometry of the protein-ligand complex and estimates the binding affinity. The two main applications of FlexX are complex prediction and virtual screening. Complex prediction is used, when you have a protein and a small molecule binding to it but no structure of the protein-ligand complex. FlexX can be used to create and rank a series of possible protein-ligand complexes. In virtual screening, you have a protein and a set of compounds and you are interested in prioritizing the compounds for experimental testing.
Other methods: Using Grid Using Graph representation END of Docking http://reco3.musc.edu/gramm/