Download

1 / 49
490 likes | 505 Views
Learn how DNA sequencing obtains the sequence of nucleotides and assembles them, overcoming challenges such as repeats, using gel electrophoresis and computational methods. Discover strategies for whole-genome sequencing.

E N D
DNA sequencing How we obtain the sequence of nucleotides of a species …ACGTGACTGAGGACCGTG CGACTGAGACTGACTGGGT CTAGCTAGACTACGTTTTA TATATATATACGTCGTCGT ACTGATGACTAGATTACAG ACTGATTTAGATACCTGAC TGATTTTAAAAAAATATT…
DNA Sequencing Goal: Find the complete sequence of A, C, G, T’s in DNA Challenge: There is no machine that takes long DNA as an input, and gives the complete sequence as output Can only sequence ~500 letters at a time
DNA Sequencing – vectors DNA Shake DNA fragments Known location (restriction site) Vector Circular genome (bacterium, plasmid) + =
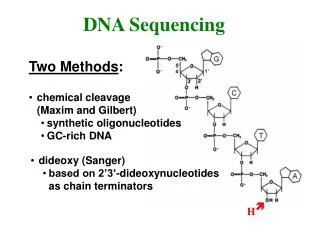
DNA Sequencing – gel electrophoresis • Start at primer (restriction site) • Grow DNA chain • Include dideoxynucleoside (modified a, c, g, t) • Stops reaction at all possible points • Separate products with length, using gel electrophoresis
Output of sequencer: a read A read: 500-700 nucleotides A C G A A T C A G …A 16 18 21 23 25 15 28 30 32 …21 Quality scores: -10log10Prob(Error) Reads can be obtained from leftmost, rightmost ends of the insert Double-barreled sequencing: (1990) Both leftmost & rightmost ends are sequenced, reads are paired
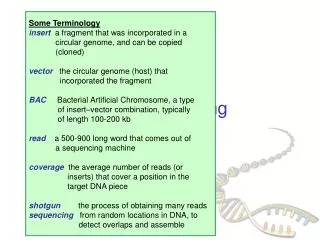
Sequencing whole genomes genome cut many times at random (Shotgun) plasmids (2 – 10 Kbp) cosmids (40 Kbp) known dist forward-reverse paired reads ~500 bp ~500 bp
Reconstructing the Sequence (Fragment Assembly) reads Cover region with ~7-fold redundancy (7X) Overlap reads and extend to reconstruct the original genomic region
Definition of Coverage C Length of genomic segment: L Number of reads: n Length of each read: l Definition:Coverage C = n l / L How much coverage is enough? Lander-Waterman model: Assuming uniform distribution of reads, C=10 results in 1 gapped region /1,000,000 nucleotides
Fragment Assembly • Computational Challenge: assemble individual short fragments (reads) into a single genomic sequence (“superstring”) • Until late 1990s the shotgun fragment assembly of human genome was viewed as intractable problem
Repeat Repeat Repeat Green and yellow fragments are interchangeable when assembling repetitive DNA Challenges in Fragment Assembly • Repeats: A major problem for fragment assembly • > 50% of human genome are repeats: - over 1 million Alu repeats (about 300 bp) - about 200,000 LINE repeats (1000 bp and longer)
Repeat Types Bacterial genomes: 5% Mammals: 50% Repeat types: • Low-Complexity DNA (e.g. ATATATATACATA…) • Microsatellite repeats (a1…ak)N where k ~ 3-6 (e.g. CAGCAGTAGCAGCACCAG) • Transposons • SINE(Short Interspersed Nuclear Elements) e.g., ALU: ~300-long, 106 copies • LINE(Long Interspersed Nuclear Elements) ~4000-long, 200,000 copies • LTRretroposons(Long Terminal Repeats (~700 bp) at each end) cousins of HIV • Gene Families genes duplicate & then diverge (paralogs) • Recent duplications ~100,000-long, very similar copies
AGTAGCACAGACTACGACGAGACGATCGTGCGAGCGACGGCGTAGTGTGCTGTACTGTCGTGTGTGTGTACTCTCCTAGTAGCACAGACTACGACGAGACGATCGTGCGAGCGACGGCGTAGTGTGCTGTACTGTCGTGTGTGTGTACTCTCCT Sequencing and Fragment Assembly 3x109 nucleotides
Strategies for whole-genome sequencing • Hierarchical –Clone-by-clone • Break genome into many long pieces • Map each long piece onto the genome • Sequence each piece with shotgun Example: Yeast, Worm, Human, Rat • Online version of (1) –Walking • Break genome into many long pieces • Start sequencing each piece with shotgun • Construct map as you go Example: Rice genome • Whole genome shotgun One large shotgun pass on the whole genome Example: Drosophila, Human (Celera), Neurospora, Mouse, Rat, Dog
Overlap-Layout-Consensus Assemblers: ARACHNE, PHRAP, CAP, TIGR, CELERA Overlap: find potentially overlapping reads Layout: merge reads into contigs and contigs into supercontigs Consensus: derive the DNA sequence and correct read errors ..ACGATTACAATAGGTT..
Finding Overlapping Reads Create local multiple alignments from the overlapping reads TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAG TTACACAGATTATTGA TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAG TTACACAGATTATTGA TAGATTACACAGATTACTGA
Find Overlapping Reads • Correcterrors using multiple alignment TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAGATTACACAGATTATTGA TAG-TTACACAGATTATTGA TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAG-TTACACAGATTACTGA TAG-TTACACAGATTATTGA insert A correlated errors— probably caused by repeats disentangle overlaps replace T with C TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA In practice, error correction removes up to 98% of the errors TAG-TTACACAGATTATTGA TAG-TTACACAGATTATTGA
Layout • Combining overlapping reads into contiguous genomic segments • Repeats are a major challenge • Do two aligned fragments really overlap, or are they from two copies of a repeat? • One solution: repeat masking – hide the repeats!!!
Merge Reads into Contigs reads contig
repeat region Merge Reads into Contigs We want to merge reads up to potential repeat boundaries Unique Contig Overcollapsed Contig
Link Contigs into Supercontigs Find all links between unique contigs Connect contigs incrementally, if 2 links supercontig (aka scaffold)
Link Contigs into Supercontigs Fill gaps in supercontigs with paths of repeat contigs
Consensus • A consensus sequence is derived from a profile of the assembled fragments • A sufficient number of reads is required to ensure a statistically significant consensus • Reading errors are corrected
Derive Consensus Sequence TAGATTACACAGATTACTGA TTGATGGCGTAA CTA TAGATTACACAGATTACTGACTTGATGGCGTAAACTA TAG TTACACAGATTATTGACTTCATGGCGTAA CTA TAGATTACACAGATTACTGACTTGATGGCGTAA CTA Derive multiple alignment from pairwise read alignments TAGATTACACAGATTACTGACTTGATGGGGTAA CTA TAGATTACACAGATTACTGACTTGATGGCGTAA CTA Derive each consensus base by weighted voting (Alternative: take maximum-quality letter)
Some Assemblers • PHRAP • Early assembler, widely used, good model of read errors • Overlap O(n2) layout (no mate pairs) consensus • Celera • First assembler to handle large genomes (fly, human, mouse) • Overlap layout consensus • Arachne • Public assembler (mouse, several fungi) • Overlap layout consensus • Phusion • Overlap clustering PHRAP assemblage consensus • Euler • Indexing Euler graph layout by picking paths consensus
A -C E -D B The Project: Building a Comparative Assembler • Assemble a closely related genome using reference genome as a guide. • Input: • Reference genome • Paired reads from unknown genome • Output: “Best” reconstruction of unknown genome Reference genome A C E B D Closely related genome
Comparative Assembly • Fragments of unknown genome: clones (100-250kb). Unknown genome 2) Sequence ends of clones (500bp). 3) Map end sequences to reference genome. x y Reference genome Each clone corresponds to pair of end sequences (ES pair)(x,y). Retain clones that correspond to a unique ES pair.
ValidES pairs • l ≤ y – x ≤ L, min (max) size of clone. • Convergent orientation. Comparative Assembly • Fragments of unknown genome: clones (100-250kb). Unknown genome 2) Sequence ends of clones (500bp). L 3) Map end sequences to reference genome. x y Reference genome
Comparative Assembly • Fragments of unknown genome: clones (100-250kb). Unknown genome 2) Sequence ends of clones (500bp). L 3) Map end sequences to reference genome. x y a b Reference genome • InvalidES pairs • Putative rearrangement in tumor • ES directions toward breakpoints (a,b): • l ≤ |x-a| + |y-b| ≤ L
x1 x2 x3 x4 y1 y2 x5 y5 y4 y3 Comparative Genome Reconstruction Reference genome (known) A C E B D Unknown sequence of rearrangements Unknown genome Map ES pairs to reference genome. Reconstruct unknown genome Location of ES pairs in reference genome. (known)
A -C E -D B x1 x2 x3 x4 y1 y2 x5 y5 y4 y3 Comparative Genome Reconstruction Reference genome (known) A C E B D Unknown sequence of rearrangements Unknown genome Map ES pairs to reference genome. Reconstruct unknown genome Location of ES pairs in reference genome. (known)
Step 1: Aligning reads to reference genome tccCAGTTATGTCAGggg Read |||||||||||| aattgccgccgtcgttttcagCAGTTATGTCAGatcttcc… Genome • Look for “best” match of read in reference genome • Not exact match Genomes not identical Sequencing errors • Can be solved in O(n2) by dynamic programming.
T GA TACA | || || TAGA TAGT Aligning Reads to Genome: Hashing • Sort all k-mers in genome (k ~ 35) • Find if read shares k-mer with genome • Extend to full alignment – throw away if not >95% similar TAGATTACACAGATTAC ||||||||||||||||| TAGATTACACAGATTAC
Step 2a: Form contigs from valid pairs L • ValidES pairs • Lmin ≤ y – x ≤ Lmax • Convergent orientation. x y Define Lmin and Lmax from length distribution of convergent clones: e.g. exclude top and bottom x% Lmin Lmax
x2 x3 x4 x1 y2 y3 y4 y1 Contigs Form groups of overlapping valid pairs
Reference Unknown genome inversion A B C A -B C t s t s translocation A B C D -C -B D A t s t s Step 2b: Invalid pairs indicate genome rearrangements • Deletion? • Insertion?
y x Clusters and Coverage Unassembled genome • Fragments of unassembled genome Rearrangement Chimeric clone 2) Sequence ends of fragments Cluster invalid pairs Isolated invalid pair 3) Map end sequences to reference genome. Reference genome
x1 x2 a y2 y1 b Clusters Clone size: (a – x1) + (b – y1) Lmin Lmax Lmax Lmin (a,b) (x1,y1) (x2,y2)
x2 x3 x4 x1 y2 y3 y4 y1 Step 3: Build Genome Graph Contigs pair of vertices with edge v w Vertex label: genome coordinates Edge label: number of pairs in contig link list of pair names
x2 x3 x4 x1 y2 y3 y4 y1 Step 3: Build Genome Graph Contigs pair of vertices with edge v w Vertex label: genome coordinates Edge label: number of pairs in contig link list of pair names
Gene2 Gene1 Step 4: Mapping Genomic Features Example: fusion genes Gene 1 Gene 2 x y a b Lmax Lmin (a,b) (x1,y1) (x2,y2) Estimate probability that gene pair (square) and breakpoint regions intersect. Respect direction of transcription
A Fusion Genome Browser Lmax Lmin (a,b) (x1,y1) (x2,y2)
Chromosomal aberrations Structural: translocations, inversions, fissions, fusions. Copy number changes: gain and loss of chromosome arms, segmental duplications/deletions. Application: Tumor Genomes Mutation and selection Compromised genome stability
Tumor Genome Architecture • What are detailed architectures of tumor genomes? • What sequence of rearrangements produce these architectures?