Download

1 / 50
680 likes | 1.31k Views
Enzyme Kinetics. Enzymes are proteins that can accelerate biochemical reactions by factors of 10 5 to 10 17 ! This is much higher than chemical catalysts. Enzymes can be extremely specific in terms of reaction substrates and products.

E N D
Enzymes are proteins that can accelerate biochemical reactions by factors of 105 to 1017! This is much higher than chemical catalysts. Enzymes can be extremely specific in terms of reaction substrates and products. Enzymes catalyze reactions under mild conditions (e.g., pH 7.4, 37ºC). The catalytic activities of many enzymes can be regulated by allosteric effectors. Enzymes Are Uniquely Powerful Catalysts
Irreversible First-Order Reactions k A B v = d[B]/dt = -d[A]/dt = k[A] (k = first-order rate constant (s-1)) Change in [A] with time (t): [A]= [A]oe–kt or [A]/[A]o = e–kt ln([A]/[A]o) = –kt ([A]o = initial concentration)
Reversible First-Order Reactions k1 A B v = -d[A]/dt = k1[A] - k-1[B] At equilibrium: k1[A]eq - k-1[B]eq = 0 [B]eq/[A]eq = k1/k-1 = Keq k-1
Second-Order Reactions k 2A P v = -d[A]/dt = k[A]2 Change in [A] with time: 1/[A] = 1/[A]o + kt A + B P v = -d[A]/dt = -d[B]/dt = k[A][B] (k = second-order rate constant (M-1s-1)) k Note: third-order reactions rare, fourth- and higher-order reactions unknown.
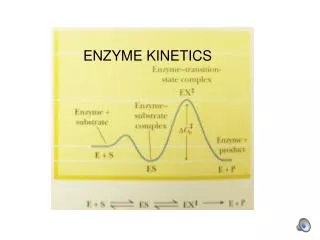
Free Energy Diagrams Keq = e–∆Gº/RT For A A‡ [A]‡/[A]o = e –∆Gº‡/RT [A]‡ = [A]o e –∆Gº‡/RT Keq = equilibrium constant [A]‡ = concentration of molecules having the activation energy [A]o = total concentration of A –∆Gº‡ = standard free energy change of activation (activation energy)
Relationship of Reaction Rate Constant to Activation Energy and Temperature: The Arrhenius Equation k = A e -Ea/RT Reaction rate constant (k) determined by activation energy (Ea or∆Gº‡, applying transition state theory) and temperature (T) and proportional to frequency of forming product (A or Q = kBT/h, where kB = Boltzmann’s constant, h = Planck’s constant): k = (kBT/h) e -G°‡/RT k =Q e-G°‡/RT G = H - T S, so: k = Q eS°‡/R e-H°‡/RT k = Q e-H°‡/RT (where Q = Q eS°‡/R) So: ln k = ln Q - H°‡/RT L-malate fumarate + H20 ln k
Relation of Equilibrium Constant to Activation Energy Keq = k1/k-1 Keq = (Q e-G1°‡/RT)/(Q e-G-1°‡/RT) Keq = e-(G1°‡ -G-1°‡)/RT ∆G° = G1°‡ - G-1°‡ Keq = e–∆G°/RT Equilibrium constant Keq says nothing about rate of reaction, only free energy difference between final and initial states. The activation energy barrier opposes reaction in both directions
Effect of a Catalyst on Activation Energy • Catalysts do not affect GA (initial) or GB (final) and so do not affect overall free energy change (∆G° = GB - GA) or equilibrium constant Keq. • Equilibrium concentrations of A and B still determined solely by overall free energy change. • Catalysts only affect ∆G°‡, lowering the activation energy. • They accelerate both the forward and reverse reaction (increase kinetic rate constants k1 and k-1).
v = k2[ES] (Note: k2 also referred to as kcat) [Enzyme]total = [E]t = [E] + [ES] How to solve for [ES]? 1. Assume equilibrium, if k-1 >> k2: KS = k-1/k1 = [E][S]/[ES] or 2. Assume steady state: d[ES]/dt = 0 Michaelis-Menten Kinetics (1) E = enzyme, S = substrate, ES = enzyme-substrate complex, P = product (Michaelis and Menten, 1913) (Briggs and Haldane, 1925)
Rate of formation of ES complex = k1[E][S] Rate of breakdown of ES complex = k-1[ES] + k2[ES] Because of steady state assumption: k1[E][S] = k-1[ES] + k2[ES] Rearranging: [ES] = (k1/(k-1 + k2))[E][S] Substituting Michaelis constant = KM = (k-1 + k2)/k1) = KS + k2/k1: [ES] = ([E][S])/KM So:KM[ES] = [E][S] Michaelis-Menten Kinetics Continued (2)
Michaelis-Menten Kinetics Continued (3) Substituting [E] = [E]t - [ES]: KM[ES] = [E]t[S] - [ES][S] Rearranging:[ES](KM + [S]) = [E]t[S] So:[ES] = [E]t[S]/(KM + [S])
Michaelis-Menten Kinetics Continued (4) Now we can substitute for [ES] in the rate equation vo = k2[ES]. But first note that thevelocity in v = k2[ES] we use is the initial velocity, vo, the velocity of the reaction after the pre-steady state and in the early part of the steady state, i.e., before ~10% of substrate is converted to product. This is because at this stage of the reaction, the steady-state assumption is reasonable ([ES] is still approximately constant). Also, since not much P has yet accumulated, we can approximate the kinetics for even reversible reactions with this equation if we limit ourselves to vo.
The Michaelis-Menten Equation vo = k2[E]t[S]/(KM + [S]) or vo = Vmax[S]/(KM + [S]) (since Vmax = k2[E]t when [S] >> KM)
E + S ES ES E + P vo = kcat[E]t[S]/(KM + [S]) Multistep Reactions k2 k3 k1 k-1 kcat = empirical rate constant that reflects rate-determining component. Mathematically, for the reaction above, kcat = k2k3/(k2 + k3). However, k2 and k3 often very hard to establish with precision as individual rate constants.
Catalytic Efficiency (kcat/KM ) “Perfect enzyme” Diffusion-controlled limit: 108-109 M-1s-1 Substrate preferences of chymotrypsin
S1 + S2 P1 + P2 A-X + B A + B-X (in transferase reactions) Sequential binding of S1 and S2 before catalysis: Random substrate binding - Either S1 or S2 can bind first, then the other binds. Ordered substrate binding - S1 must bind before S2. Ping Pong reaction - first S1 P1, P1 released before S2 binds, then S2 P2. Bisubstrate Reactions E E
Sequential binding Ternary complex Ping Pong reaction
Indicative of ternary complex formation and a sequential mechanism
Reversible inhibition (Inhibitors that can reversibly bind and dissociate from enzyme; activity of enzyme recovers when inhibitor diluted out; usually non-covalent interaction.) Competitive Mixed (noncompetitive) Uncompetitive Irreversible inhibition (Inactivators that irreversibly associate with enzyme; activity of enzyme does not recover with dilution; usually covalent interaction.) Types of Enzyme Inhibition
Effects of Competitive Inhibitor on Enzyme Kinetics KI (inhibitor dissociation constant)=koff/kon KappM = KM(1 + [I]/KI)> KM Vappmax = Vmax
For a competitive inhibitor of an enzyme that follows Michaelis-Menton kinetics: vI/v0 = (Vmax[S]/(KMa + [S]))/(Vmax[S]/(KM + [S])) = (KM + [S])/(KMa + [S]) vI = initial velocity with inhibitor v0 = initial velocity without inhibitor a = 1 + [I]/KI When vI/v0= 0.5, [I] = IC50 = KI(1 + [S]/KM) If measurement made when [S] << KM, IC50 = KI Relationship of KI to Half-Maximal Inhibitory Concentration (IC50)
Effects of Uncompetitive Inhibitor on Enzyme Kinetics KappM = KM/(1 + [I]/KI)< KM Vappmax = Vmax/(1 + [I]/KI)< Vmax
Effects of Mixed (Noncompetitive) Inhibitor on Enzyme Kinetics • Not the same as uncompetitive inhibition. • In mixed inhibition, inhibitor can bind E or ES. k1 k-1 KappM = (1 + [I]/KI)KM/(1 + [I]/KI) (= KM, when KI = KI, which is often the case.) Vappmax = Vmax/(1 + [I]/KI) < Vmax
a = 1 + [I]/KI a = 1 + [I]/KI
(For mixed inhibitor, generally, ~ KM) a = 1 + [I]/KI a = 1 + [I]/KI
Irreversible Inhibition k1 k2 E + I E·I E-I Plot: ln(residual enzyme activity) vs. time If [I]>>[E], conditions are pseudo-first order and slope is -kobs (pseudo-first order inactivation rate constant) kinact (second-order inactivation constant) = k1k2/k-1 = kobs/[I] k-1 Slope = -kobs
Irreversible Inhibition by Adduct Formation (diisopropylfluorophosphate)
Irreversible Inhibition of Chymotrypsin by TPCK (N-tosyl-L-phenylalanine chloromethylketone)