Download

1 / 33
330 likes | 579 Views
一、 PCR 的定义: PCR ( polymerase chain reaction) : 聚合酶链式反应,又称体外 DNA 扩增技术。 近年来发展起来的一种体外扩增特异 DNA 片段的技术。. DNA 扩增的传统方法: 一般采用分子克隆法,需将构建的含有目的基因的载体导入细胞进行扩增,并需要用探针进行筛选,牵扯到 DNA 酶切、连接、转化、培养及探针杂交等技术,虽然技术上已无难点,但 操作复杂,需数周到数月的时间,且不利于普及 。.

E N D
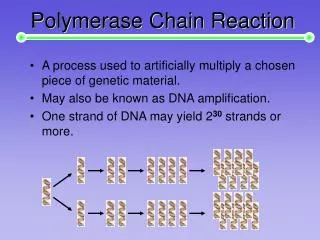
一、PCR的定义: PCR(polymerase chain reaction) : 聚合酶链式反应,又称体外DNA扩增技术。 近年来发展起来的一种体外扩增特异DNA片段的技术。
DNA扩增的传统方法: 一般采用分子克隆法,需将构建的含有目的基因的载体导入细胞进行扩增,并需要用探针进行筛选,牵扯到DNA酶切、连接、转化、培养及探针杂交等技术,虽然技术上已无难点,但操作复杂,需数周到数月的时间,且不利于普及。
PCR技术:1985年由美国 Cetus 公司的Kary Mullis首创,可以将微量目的DNA片段扩增一百万倍以上。 优点:敏感度高、特异性强、产率高、重复性好以及快速简便等,广泛应用于微生物学、考古学、法医学及体育等领域,并已普及到许多普通实验室,大大简化了传统的分子克隆技术,从而比较容易地对目的基因进行分析、鉴定。 Kary Mullis本人因此获1993年诺贝尔化学奖。
二、PCR的原理: 用于扩增位于两段已知序列之间的DNA片段,类似于天然DNA的复制过程。 以拟扩增的DNA分子为模板,以一对分别与模板5′末端和3′末端互补的寡核苷酸片段为引物,在DNA聚合酶的作用下,按照半保留复制的机制沿着模板链延伸直至完成新的DNA合成,重复这一过程,即可使目的DNA片段得到扩增。
扩增的特异性取决于引物与模板DNA的特异结合,基本反应步骤分三步:扩增的特异性取决于引物与模板DNA的特异结合,基本反应步骤分三步: 1. 变性(Denaturation): 加热使模板DNA双链间的氢键断裂而形成两条单链。94℃ 30″ 2. 退火 (复性) (Annealling): 突然降温后模板DNA与引物按碱基配对原则互补结合,也存在两条模板链之间的结合,但由于引物的高浓度,结构简单的特点,主要的结合发生在模板与引物之间。55 ℃ 30 ″
3. 延伸(Extension): 将反应温度调节到酶的最适温度,在DNA聚合酶、4种 dNTPs 及镁离子等存在的条件下,以引物的 3′端开始,结合单核苷酸,形成与模板链互补的新DNA链。72 ℃ 1′ 上述 3 步为一个循环,每经过一个循环,样本中的DNA量应该增加一倍,新形成的链又可成为新一轮循环的模板,经过25~40个循环后DNA可扩增106 ~ 109 倍。
PCR 的基本反应步骤 变性 95˚C 退火 Tm-5˚C 延伸 72˚C
PCR扩增的特异性是由一对寡核苷酸引物所决定的。反应初期,原来的DNA担负起使模板的作用,随着循环次数的递增,由引物介导延伸的片段急剧增多而成为主要模板,最终的扩增产物是介于两种引物5’端之间的DNA片段。PCR扩增的特异性是由一对寡核苷酸引物所决定的。反应初期,原来的DNA担负起使模板的作用,随着循环次数的递增,由引物介导延伸的片段急剧增多而成为主要模板,最终的扩增产物是介于两种引物5’端之间的DNA片段。
三、PCR的成分和作用:终浓度 1. 缓冲液: 10~ 50 mM Tris- Cl (pH8.4) 维持 Taq 酶作用环境的偏碱性 25 ~ 50 mM KCl 促进引物退火,>50 mM 会抑制 Taq 酶的活性。 100μg / ml 牛血清白蛋白(BSA) 对酶有一定的保护性,如质量不好将起相反的作 用,建议使用乙酰化的 BSA。明胶、Tween-20、 二硫苏糖醇 (DTT) 也有类似作用。
2. MgCl2: 1.5~ 2.0 mM Taq 酶具有 Mg2+依赖性,显著影响反应的特异性和扩增片段的产量,过量能增加非特异扩增并影响产率,过低则酶活性显著下降。
3. dNTPs : dATP、 dGTP 、dCTP、dTTP——底物 0.02 ~ 0.2 mM dNTPs 可与 Mg2+ 结合,应注意Mg2+ 浓度与dNTPs 浓度之间的关系, Mg2+ 浓度比 dNTPs 浓度高 0.2 ~ 2.5 mM。 过高:加快反应速度,还可增加碱基的错误掺入 率和室验成本。 过低:反应速度下降,可提高实验的精确性。
4. 引物 (Primer-P): 预扩增核酸片段两端的已知序列,决定特异性。 0.2 ~ 1 μM 偏高:非特异产物扩增及错配,增加引物之间 形成引物二聚体,产量降低。 偏低:产量降低。
5. Taq DNA 聚合酶:耐高热 0.5 ~ 5 U / 100 μl 1U / 25 ~ 50μl 偏高:引物非特异产物的扩增 偏低:产物量降低
6. 模板DNA (Template): 最低102~ 105 bp DNA片段,实际用量远远 超过此量,用量需在实验中摸索。1 ~ 5 μl 过高:非特异产物增加 过低:产量降低 7. 水: 去离子水,补足整个反应体积。
四、PCR反应体系: 常用30μl 各种成分的实际用量应根据实验者选用的该成分的终浓度及所拥有的储备液浓度进行核算。
10×buffer: 1×30 / 10 = 3μl MgCl2: 1.5×30 / 25 = 1.8μl dNTPs: 0.2×30 / 10 = 0.6μl 引物 P: 0.4×30×2 / 10 = 2.4μl Taq 酶(5U/μl):1 / 5 = 0.2μl 模板: 1μl 水: 30-3-1.8-0.6-2.4-0.2-1=21μl
操作:5份标本 总体积 30μl ×6 = 180μl 1. 取一 0.5 ml Ep 管,依次加入下列试剂: 10×buffer: 3μl ×6 = 18μl MgCl2: 1.8μl×6 = 10.8μl dNTPs: 0.6μl×6 = 3.6μl 引物 P: 2.4μl ×6 = 14.4μl Taq 酶 (5U/μl): 0.2μl × 6 = 1.2μl 水: 21μl × 6 = 126μl 共174μl,先用移液器 / 指弹混匀,后用离心机瞬时 混匀(管壁上液体沉至管底)。
2. 另取 5 支0.2ml Ep管,分别加入上述液体29μl。 3. 分别取标本模板各1μl,加入上述5支 Ep 管中,混匀,分别加入2滴液体石蜡(防止加温过程中液体蒸发影响反应体积),离心机瞬时离心,备用。 4. 反应条件:PCR扩增仪 94℃30″,55 ℃ 30 ″,72 ℃ 1′, 35个循环,72 ℃延伸 5 ′。
五、PCR反应条件优化: 1、温度、循环参数: ⑴ 变性温度和时间: 保证模板DNA解链完全是保证整个PCR扩增成功的关键。 加热90~95℃,30 ~ 60 s,再复杂的DNA分子也可变性为单链。根据模板DNA复杂程度,可以调整变性温度和时间。一般情况下选择94 ℃ 30 ″,可使各种复杂的DNA分子完全变性。 温度过高或高温持续时间过长,可对Taq酶活性和dNTP分子造成损害。
⑵ 复性温度和时间: PCR扩增特异性取决于复性过程中引物与模板 的结合。 复性温度的选择,可根据引物的长度和G+C含量确定,长度在15 ~ 25 bp 之间时,复性温度 Tm = 4 (G+C) +2 (A+T) 计算得到,一般位于40 ~ 60℃,30 ~ 60 s。 复性温度越高,产物特异性越高。复性温度越低,产物特异性越低。 一般情况下选择55 ℃ 30″,足以使引物与模板之间完全结合。
⑶ 延伸温度和时间: 一般位于Taq酶最适作用温度70 ~ 75 ℃之间。 引物小于16个核苷酸时,过高的延伸温度不利于引 物与模板的结合,可以缓慢升温到70 ~ 75 ℃。 延伸反应时间,可根据待扩增片段的长度而定,< 1Kb,1分钟足够;> 1Kb需加长延伸时间,10Kb片段延伸时间可达15分钟。 延伸时间过长可出现非特异扩增,常用72 ℃ 1′。
⑷ 循环数: 其他参数选定后,PCR循环次数主要取决于模板DNA的浓度。 理论上说20 ~ 25次循环后,PCR产物的积累即可达到最大值,实际操作中由于每步反应的产率不可能达到100%,因此不管模板浓度是多少,20 ~ 30次是比较合理的循环次数。 循环次数越多,非特异扩增增加。
2、MgCl2浓度: 可显著影响PCR的产量及产物特异性 1.5~ 2.0 mM 过高:增加非特异扩增并影响产率 过低:酶活性显著下降。
3、引物: 0.2 ~ 1 μM 偏高:非特异产物扩增及错配,增加引物 之间形成引物二聚体,产量降低。 偏低:产量降低。 引物设计原则: 总原则是提高扩增的效率和特异性
引物设计原则 ⑴ 长度15~30 b,过短特异性降低,过长则成本增加,产量也下降。 ⑵ 引物碱基尽可能随机分布,避免出现嘌呤、嘧啶堆积现象,引物 G+C 含量宜在45 ~ 55%左右。 ⑶ 引物内部不能含有自身互补序列,否则会形成发夹样二级结构,两个引物之间尤其在 3’ 末端不应有互补链存在,不然会形成引物二聚体。
引物设计原则 ⑷ 引物的碱基顺序不应与非扩增区域有同源性(<70%)或少于连续8个的互补碱基。要求在引物设计时采用计算机进行辅助检索分析。 ⑸ 引物的 5′末端碱基:并没有严格的限制,只要与模板DNA结合的引物长度足够,其 5′ 末端碱基可以不与模板DNA匹配呈游离状态。最多可游离10个碱基并不影响PCR反应进行。
引物设计原则 ⑹ 引物的 3′末端碱基:原则上要求引物的3′末端与模板DNA一定要配对,另外 3′末端的末位碱基在很大程度上影响着 Taq 酶的延伸效率。 引物3′末端碱基在错误配对时不同碱基的引发效率存在很大差异,最好选T、G、C,而不选A。
六、PCR扩增产物的分析 1、凝胶电泳分析法: 聚丙烯酰胺凝胶电泳灵敏度远高于琼脂糖电泳, 用于探讨PCR扩增的灵敏度效果好,但配制复杂, 常用琼脂糖凝胶电泳。 2、点杂交 3、微孔板杂交法
七、PCR技术的主要用途 1、目的基因的克隆: PCR技术为在重组DNA过程中获得目的基 因片段提供了简便快速的方法。 2、基因的体外突变: 可以随意设计引物在体外对目的基因片段 进行嵌和、缺失、点突变等改造。
3、DNA和RNA的微量分析: PCR技术高度敏感,对模板DNA的含量 要求很低,是DNA和RNA微量分析的最好方 法。实际工作中,一滴血液、一根毛发或一个细 胞已足以满足PCR的检测需要,因此在基因诊 断方面具有极广阔的应用前景。 4、DNA序列测定: PCR技术的引入使DNA测序工作大大简化, 也提高了测序的速度。 5、基因突变分析: 利用PCR与一些技术的结合可以大大提高 基因突变检测的敏感性。