Download
1 / 1
10 likes | 197 Views
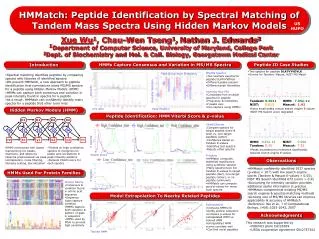
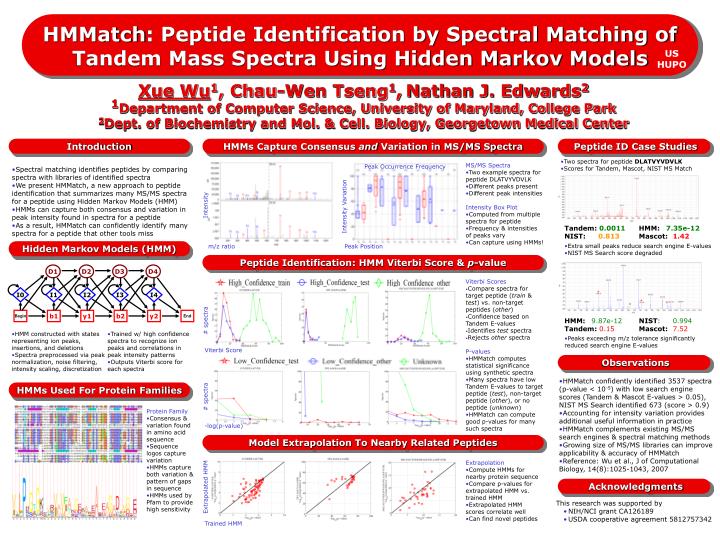
D1. D2. D3. D4. I0. I1. I2. I4. I3. Begin. b1. y1. b2. y2. End. HMMatch : Peptide Identification by Spectral Matching of Tandem Mass Spectra Using Hidden Markov Models. US HUPO.

E N D
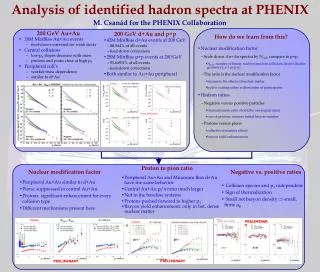
D1 D2 D3 D4 I0 I1 I2 I4 I3 Begin b1 y1 b2 y2 End HMMatch: Peptide Identification by Spectral Matching of Tandem Mass Spectra Using Hidden Markov Models US HUPO Xue Wu1, Chau-Wen Tseng1,Nathan J. Edwards21Department of Computer Science, University of Maryland, College Park2Dept. of Biochemistry and Mol. & Cell. Biology, Georgetown Medical Center Introduction HMMs Capture Consensus and Variation in MS/MS Spectra Peptide ID Case Studies • Two spectra for peptide DLATVYVDVLK • Scores for Tandem, Mascot, NIST MS Match Intensity • MS/MS Spectra • Two example spectra for peptide DLATVYVDVLK • Different peaks present • Different peak intensities • Intensity Box Plot • Computed from multiple spectra for peptide • Frequency & intensities of peaks vary • Can capture using HMMs! Peak Occurrence Frequency • Spectral matching identifies peptides by comparing spectra with libraries of identified spectra • We present HMMatch, a new approach to peptide identification that summarizes many MS/MS spectra for a peptide using Hidden Markov Models (HMM) • HMMs can capture both consensus and variation in peak intensity found in spectra for a peptide • As a result, HMMatch can confidently identify many spectra for a peptide that other tools miss Intensity Variation • Tandem: 0.0011 HMM: 7.35e-12 • NIST: 0.813 Mascot: 1.42 • Extra small peaks reduce search engine E-values • NIST MS Search score degraded Hidden Markov Models (HMM) m/z ratio Peak Position Peptide Identification: HMM Viterbi Score & p-value • Viterbi Scores • Compare spectra for target peptide (train & test) vs. non-target peptides (other) • Confidence based on Tandem E-values • Identifies test spectra • Rejects other spectra # spectra • HMM:9.87e-12 NIST: 0.994 • Tandem: 0.15Mascot:7.52 • Peaks exceeding m/z tolerance significantly reduced search engine E-values • HMM constructed with states representing ion peaks, insertions, and deletions • Spectra preprocessed via peak normalization, noise filtering, intensity scaling, discretization • Trained w/ high confidence spectra to recognize ion peaks and correlations in peak intensity patterns • Outputs Viterbi score for each spectra Viterbi Score • P-values • HMMatch computes statistical significance using synthetic spectra • Many spectra have low Tandem E-values to target peptide (test), non-target peptide (other), or no peptide (unknown) • HMMatch can compute good p-values for many such spectra Observations • HMMatch confidently identified 3537 spectra • (p-value < 10-5) with low search engine scores (Tandem & Mascot E-values > 0.05), • NIST MS Search identified 673 (score > 0.9) • Accounting for intensity variation provides additional useful information in practice • HMMatch complements existing MS/MS search engines & spectral matching methods • Growing size of MS/MS libraries can improve applicability & accuracy of HMMatch • Reference: Wu et al., J of Computational Biology, 14(8):1025-1043, 2007 # spectra HMMs Used For Protein Families • Protein Family • Consensus & variation found in amino acid sequence • Sequence logos capture variation • HMMs capture both variation & pattern of gaps in sequence • HMMs used by Pfam to provide high sensitivity -log(p-value) Model Extrapolation To Nearby Related Peptides Extrapolated HMM • Extrapolation • Compute HMMs for nearby protein sequence • Compare p-values for extrapolated HMM vs. trained HMM • Extrapolated HMM scores correlate well • Can find novel peptides Acknowledgments • This research was supported by • NIH/NCI grant CA126189 • USDA cooperative agreement 5812757342 Trained HMM