Download
1 / 50
530 likes | 836 Views
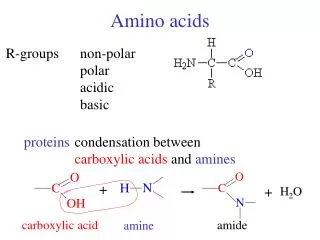
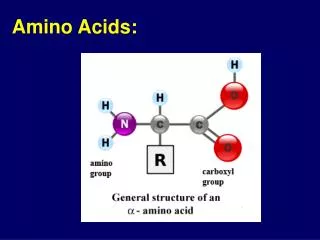
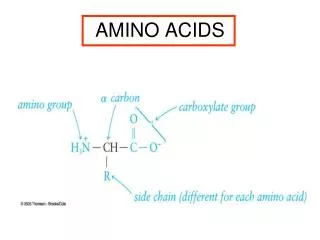
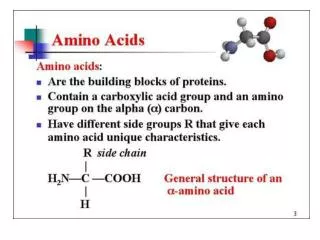
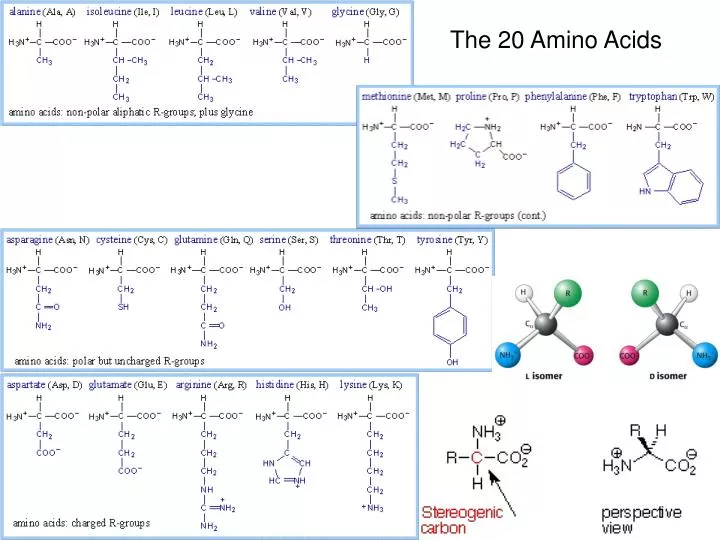
The 20 Amino Acids. Amino Acid Similarities. buried vs. exposed: compare average buried surface area or solvent-exposure correlation with partition coefficient and free-energy of transfer hydrophobicity scales... AAindex: online database http://www.genome.jp/aaindex/

E N D
Amino Acid Similarities • buried vs. exposed: compare average buried surface area or solvent-exposure • correlation with partition coefficient and free-energy of transfer • hydrophobicity scales... • AAindex: online database • http://www.genome.jp/aaindex/ • thousands of dimensions of similarity • Cornette et al. (1987) – factor analysis • Janin (1979)1; (2) Wolfenden, et al.2; • (3) Kyte and Doolittle 3; and (4) Rose, et al
Rose (1985). Science. Richards (1977) Ann. Rev Biophys Bioeng
Evolution reflects a combination of multiple similarities... PAM250 (Dayhoff, 1978) – 10 x log_odds of substitution (relative to rate expected from random)
Bond lengths 3.4A
steric conflicts trans eclipsed gauche (+/- 60 deg torsion angles)
2 degrees of freedom per AA in backbone left-handed helices beta-sheets right-handed-alpha-helices Ramachandaran Plots
both forms have clashes, so there is less preference (cis allowed) ~5% cis (~2 kcal/mol delta G) 50% of proteins have a cis peptide bond, 87% are Pro 0.002/sec flipping frequency (~10 min) (14-24 kcal/mol activation barrier) role in turns Pal & Chakrabarti (1999), JMB 5-HT neurotransmitter receptor/ion channel (Dougherty, Nature, 2005) Kun Ping Lu1, Greg Finn1, Tae Ho Lee1 & Linda K Nicholson (2007). Prolyl cis-trans isomerization as a molecular timer. Nature Chemical Biology 3, 619 – 629.
Rotamers • libraries • Ponder and Richards • Dunbrack • Richardson • chi-angles • conflict with carbonyl O (show picture) • backbone-independent and –dependence (conditional)
Tuffery et al. • http://bioserv.rpbs.jussieu.fr/doc/Rotamers.html aa chi1 chi2 num freq std.dev. -- ---- ---- --- ---- -------- TYR -67.50 82.50 5364 0.2326 12.1920 TYR -62.50 -77.50 5262 0.2282 8.8149 TYR -62.50 -22.50 1442 0.0625 12.1077 TYR 62.50 -82.50 1122 0.0487 8.3048 TYR 62.50 82.50 1618 0.0702 8.0934 TYR -177.50 -82.50 1522 0.0660 11.8303 TYR 177.50 77.50 6728 0.2918 10.0764 VAL -62.50 4225 0.1843 15.1085 VAL 67.50 1941 0.0847 20.3667 VAL 177.50 16754 0.7310 9.8021
Dunbrack & Cohen: BBDep library • Bayesian statistics • counts in bins -> conditional prob’s • use Dirichlet priors • infer posteriors by simulation
Disulfide bridges • only non-linear connection; adds stability • intracellular environment is usually reducing • secreted proteins have disulfides bridges more often • dsbABCD – disulfide-bond isomerases • glutathione reductases
disulfide conformations • Ca-Ca dist: 4.5-7.5Å • Richardson (1981) • left-handed spiral, right-handed hook • adjacent Cys: (thioredoxin), near: Zn-finger (C-X-X-C) • buried vs. solvent-exposed • more prevalent in secreted proteins (immunoglobulins, chymotrypsin, insulin...); cytosol is usually a reducing environment
Insulin Immunoglobulins (IgG)
Contribution of disulfides to protein stability • Can you increase stability by engineering in a disulfide? • Betz (1993) Protein Science • effects on DH vs. DS: main effect comes from reducing entropy of unfolded state • disruption of Cys6-Cys127 in HEL lysozyme costs 7.5 kcal/mol • disruption of 1 disulfide in RNase T1 costs 3.3 kcal/mol • disruption of Cys14-Cys38 in BPTI costs 8 kcal/mol
Alpha-helices • standard alpha-helices (predominant form) • right-handed • H-bonds: i:i+4 • O points forward • 3.6 residues per turn of helix (100 deg/aa) • p-helices (i:i+5) • tighter, examples? • often near ends? • 3/10-helices (i:i+3) 1EHK, 153-157, chain B • left-handed helices • Ramachandaran plot “disallowed” region • examples: alanine racemase (res 40-44), nitrate reductase, collagen • 87% are short (only 4 residues long) • Novotny and Kleywegt (2005)
p helix (i:i+5) 3/10 helix (i:i+3) a helix (i:i+4) carbonyl oxygens point forward Cb’s point slightly backward
Helical Trivia • helix dipole • C-cap (JMB paper) • http://dx.doi.org/10.1016/S0022-2836(02)00734-9 • N-cap: Ser, Thr • helix packing angles (Bowie, 1997) • helix bundles, hemoglobin, leu-zippers • kinks: Pro (disrupt H-bonds), see 1MLT
Beta-sheets • parallel • anti-parallel • twist • ladder of H-bonds • side-chains alternate up and down (pleated) • topology, Greek keys (5PCY), jelly rolls • beta-bulge (RNase A, 1Z6S, res 88-91) nitrate reductase
antiparallel parallel • good examples to look at: • flavodoxin (1CZN) – 5-stranded parallel • immunoglobulin (4FAB) - antiparallel • see twist of sheet in 2o2v (kinase) • notice C=O and Ca-Cb vectors, H-bonds, twist of sheet
Turns • defined when C-alpha atoms are < 7A apart • A γ-turn is characterized by hydrogen bond(s) in which the donor and acceptor residues are separated by two residues (i:i+2). • A β-turn (the most common form) is characterized by hydrogen bond(s) in which the donor and acceptor residues are separated by three residues (i:i+3). • An α-turn is characterized by hydrogen bond(s) in which the donor and acceptor residues are separated by four residues (i:i+4). • A π-turn is characterized by hydrogen bond(s) in which the donor and acceptor residues are separated by five residues (i:i+5). • An ω-loop is a catch-all term for a longer loop with no internal hydrogen bonding. • role of Gly, Pro... • Richardson, 1980; Wilmot and Thornton, 1988
Residue 2 Residue 3 • Designation Phi,Psi Phi,Psi Comments • ----------- ---------- --------- -------- • I -60,-30 -90,0 Most common type. • II -60,120 80,0 • III -60,-30 -60,-30 Like 3/10 helix. • IV unclassified turns • V -80,80 80,-80 • VIa -60,120 -90,0 * • VIb -120,120 -60,0 * • VII** • VIII -60,-30 -120,120 • --------------------------------------------------------------- • * 2-3 peptide bond is cis, residue 3 is proline. • ** Type VII is a bend recognized by psi(2)~180 and phi(3)<60 or by psi(2)<60 and phi(3)~180. • Favorable and (unfavorable) Residues in Beta-turns by Position: • Type Residue 1 Residue 2 Residue 3 Residue 4 • ----- --------- --------- --------- ----------- • I AspAsnSerCys Pro (Pro) Gly • II AspAsnSerCys Pro Gly, Asn Gly • VIa Pro • VIb Pro http://www.bmb.uga.edu/wampler/tutorial/prot2.html
secondary structure length distributions means: alpha-helices: ? beta-sheets: ?
DSSP • Kabsch and Sander (1983) • secondary structure identification based on geometry (f/y angles) AND H-bonding patterns • identifies sub-types of helices, turns, etc. • calculates solvent accessibility • patterns and merging rules H-bond criteria: up to 5A or 60º (but not both)
# RESIDUE AA STRUCTURE BP1 BP2 ACC N-H-->O O-->H-N N-H-->O O-->H-N TCO KAPPA ALPHA PHI PSI X-CA Y-CA Z-CA 1 2 A T 0 0 47 0, 0.0 728,-1.9 0, 0.0 2,-0.4 0.000 360.0 360.0 360.0 123.8 39.4 15.0 22.0 2 3 A L - 0 0 88 725,-0.2 2,-0.4 726,-0.2 725,-0.2 -0.962 360.0-170.9-117.7 126.1 36.4 12.8 22.6 3 4 A L B -A 726 0A 3 723,-2.9 723,-3.0 -2,-0.4 8,-0.1 -0.944 7.9-152.7-117.8 141.8 32.8 13.9 22.1 4 5 A G - 0 0 2 -2,-0.4 2,-0.5 721,-0.2 7,-0.2 -0.182 31.3 -88.3 -94.2-165.9 29.7 11.9 23.1 5 6 A T > - 0 0 4 719,-0.3 3,-2.1 740,-0.1 6,-0.3 -0.920 45.7-105.5-108.1 127.9 26.2 12.0 21.6 6 7 A A T 3 S+ 0 0 1 28,-1.6 29,-0.1 -2,-0.5 7,-0.1 -0.224 103.8 14.8 -54.2 136.7 23.7 14.5 23.0 7 8 A L T 3 S+ 0 0 51 1,-0.3 -1,-0.2 5,-0.1 28,-0.0 0.266 104.9 104.3 84.2 -9.1 21.1 12.9 25.2 8 9 A R S X S- 0 0 60 -3,-2.1 3,-2.3 716,-0.1 -1,-0.3 -0.606 88.0-100.3 -97.2 162.2 23.0 9.6 25.5 9 10 A P T 3 S+ 0 0 100 0, 0.0 714,-0.1 0, 0.0 -2,-0.1 0.801 122.0 47.9 -52.9 -37.5 24.9 8.6 28.6 10 11 A A T 3 S+ 0 0 37 -5,-0.1 -6,-0.2 714,-0.1 -4,-0.1 0.294 86.0 145.7 -89.6 12.1 28.3 9.5 27.3 11 12 A A < - 0 0 2 -3,-2.3 2,-0.6 -6,-0.3 -5,-0.1 -0.021 51.7-126.2 -49.0 148.5 27.0 12.9 26.2 12 13 A T - 0 0 15 22,-0.4 24,-2.9 -8,-0.1 2,-0.5 -0.885 31.5-152.1 -97.5 120.9 29.2 16.0 26.3 13 14 A R E -c 36 0B 47 -2,-0.6 63,-2.9 61,-0.2 64,-1.8 -0.905 15.0-172.4-110.2 129.7 27.3 18.7 28.2 14 15 A V E -cd 37 77B 0 22,-2.8 24,-2.7 -2,-0.5 2,-0.5 -0.957 8.6-156.2-115.6 127.9 27.6 22.4 27.8 15 16 A M E -cd 38 78B 0 62,-3.4 64,-4.0 -2,-0.4 2,-0.6 -0.927 6.8-153.5-107.5 123.8 25.9 24.9 30.2 16 17 A L E -cd 39 79B 2 22,-2.8 24,-2.8 -2,-0.5 2,-1.0 -0.891 2.3-160.8 -98.4 119.4 25.2 28.4 28.8 17 18 A L E S+cd 40 80B 0 62,-1.9 64,-3.6 -2,-0.6 65,-0.8 -0.841 80.7 24.8-100.8 90.1 25.1 31.1 31.5 18 19 A G - 0 0 0 22,-2.7 23,-0.2 -2,-1.0 22,-0.1 0.221 67.5-156.8 113.4 119.6 23.2 33.7 29.5 19 20 A S + 0 0 0 20,-0.1 29,-2.6 4,-0.1 30,-0.5 -0.240 44.5 129.7-117.7 37.4 21.0 32.7 26.6 20 21 A G S > S- 0 0 6 27,-0.2 4,-2.2 26,-0.1 28,-0.3 0.001 81.7 -72.1 -78.5-168.2 20.9 36.0 24.5 21 22 A E H > S+ 0 0 15 1,-0.2 4,-1.1 2,-0.2 5,-0.1 0.737 134.8 53.5 -59.0 -30.3 21.6 36.2 20.7 22 23 A L H > S+ 0 0 23 2,-0.2 4,-1.4 1,-0.2 3,-0.4 0.932 111.1 44.4 -71.6 -46.8 25.3 35.6 21.3 23 24 A G H > S+ 0 0 3 1,-0.2 4,-2.9 2,-0.2 -2,-0.2 0.865 105.2 65.8 -63.1 -35.2 24.6 32.4 23.3 24 25 A K H X S+ 0 0 6 -4,-2.2 4,-1.9 1,-0.2 -1,-0.2 0.880 103.7 44.3 -54.5 -42.6 22.1 31.3 20.7 25 26 A E H X S+ 0 0 10 -4,-1.1 4,-2.4 -3,-0.4 -1,-0.2 0.830 110.9 53.5 -73.8 -32.9 24.8 31.0 18.1 26 27 A V H X S+ 0 0 5 -4,-1.4 4,-2.1 2,-0.2 -2,-0.2 0.934 108.8 51.3 -65.7 -41.5 27.1 29.2 20.5 27 28 A A H X S+ 0 0 0 -4,-2.9 4,-3.0 2,-0.2 -2,-0.2 0.923 109.8 49.3 -58.2 -46.8 24.1 26.7 21.1 28 29 A I H X S+ 0 0 0 -4,-1.9 4,-2.0 1,-0.2 -1,-0.2 0.941 110.9 48.7 -61.1 -46.3 23.8 26.2 17.4 29 30 A E H < S+ 0 0 22 -4,-2.4 4,-0.3 2,-0.2 -1,-0.2 0.840 112.4 48.6 -64.1 -31.7 27.4 25.5 17.0 30 31 A C H ><>S+ 0 0 0 -4,-2.1 5,-2.3 1,-0.2 3,-1.8 0.940 110.8 50.6 -69.9 -47.3 27.3 23.1 19.9 31 32 A Q H ><5S+ 0 0 0 -4,-3.0 3,-1.6 1,-0.3 -2,-0.2 0.818 100.7 63.6 -58.1 -31.6 24.3 21.3 18.5 32 33 A R T 3<5S+ 0 0 22 -4,-2.0 709,-1.8 1,-0.3 -1,-0.3 0.664 106.7 44.6 -68.2 -15.9 26.1 21.0 15.1 33 34 A L T < 5S- 0 0 21 -3,-1.8 -1,-0.3 -4,-0.3 -2,-0.2 0.162 122.1-106.9-108.1 8.2 28.7 18.8 16.9 34 35 A G T < 5 + 0 0 0 -3,-1.6 -28,-1.6 1,-0.2 -22,-0.4 0.770 66.7 158.0 68.3 25.3 26.0 16.8 18.7 35 36 A V < - 0 0 0 -5,-2.3 2,-0.3 -30,-0.1 -1,-0.2 -0.605 45.0-118.9 -82.2 134.6 26.9 18.6 22.0 36 37 A E E -c 13 0B 11 -24,-2.9 -22,-2.8 -2,-0.3 2,-0.5 -0.628 32.2-156.1 -79.5 135.4 24.2 18.5 24.6 37 38 A V E -c 14 0B 0 -2,-0.3 17,-1.7 -24,-0.2 16,-1.5 -0.941 19.4-176.2-125.0 125.0 23.1 22.1 25.6 38 39 A I E -ce 15 54B 0 -24,-2.7 -22,-2.8 -2,-0.5 2,-0.5 -0.957 19.2-155.4-114.6 115.4 21.5 23.4 28.7 39 40 A A E -ce 16 55B 0 15,-2.5 17,-2.0 -2,-0.6 2,-0.4 -0.847 11.4-174.1-102.5 130.5 20.6 27.1 28.4
H = alpha helix B = residue in isolated beta-bridge E = extended strand, participates in beta ladder G = 3-helix (3/10 helix) I = 5 helix (pi helix) T = hydrogen bonded turn S = bend (direction change by > 70 degrees)
identical peptide fragments up to 8-mers can be found in both alpha-helical and beta-strand conformations in different proteins/contexts • Zhou, F. Alber, G. Folkers, G. Gonnet and Chelvanayagam (2000) • Design of Protein Conformational Switches • Ambroggio and Brian Kuhlman (2006) • discusses how to engineer regions that can change states • also discusses relation of alternative folding states to amyloid formation • Chameleon peptides (Minor & Kim, Nature, 1996) • 11-mer as both a-helix and b-strand in GB1
Secondary Structure Prediction • Chou/Fasman • aa propensities • alpha-helix preference (aliphatic, non-branched): • Ala, Leu, Met, Phe, Glu, Gln, His, Lys, Arg • beta-sheet preference (hydrophobic): • Tyr, Trp, (Phe, Met), Ile, Val, Thr, Cys • rules: nucleation, helix-breaker • 60-70% accuracy • a helix is predicted if, in a run of six residues, four are helix favoring and the average valued of the helix propensity is greater than 1.0 and greater than the average strand propensity. Such a helix is extended along the sequence until a proline is encountered (helix breaker) or a run of 4 residues with helical propensity less than 1.0 is found. • A strand is predicted if, in a run of 5 residues, three are strand favouring, and the average value of the strand propensity is greater than 1.04 and greater than the average helix propensity. Such a strand is extended along the sequence until a run of 4 residues with strand propensity less than 1.0 is found.
secondary structure propensities (relative to overall frequency)
PHD (Rost and Sander, 1993) • exploits evolutionary information (multiple alignment of family members) • neural network, window-size=17 residues • 70-75% accuracy • limits of prediction? (rest is due to non-local interactions) • identical fragments up to 7 aa can be found in both helices and sheets
Transmembrane regions • single helix (endolysins) • helix bundle (6-12) (K+ channel, ABC transporters, GPCRs) • beta barrel (OMP)
Predicting Transmembrane Regions • hydrophobic (no moment) • charged near ends (like membrane) • positive-inside rule (von Heinje, 1992) • characteristic lengths (15-35 for helices, with caps) • TMPred • TMMHM (Sonnhammer) gluconate permease 3 from E. coli
Signal Peptides • SignalP (Nielsen, von Heinje, Brunak) - HMM • signal peptidases (gram-pos. vs. gram-neg. vs. eukaryotic) • isoelectric point differences • The average length of signal peptides range from 22 (eukaryotes) and 24 (Gram-negatives) to 32 amino acid residues for Gram-positives (-3,-1) rule – small and neutral
Disordered regions • Keith Dunker group • a 4th category of secondary structure • not just random coil (which has irregular but fixed f/y, H-bonds) • unstructured, molten globule, meta-stable • flexible, dynamic, sample multiple conformations in solution (HSQC) • correlation with B-factors, dis-order in crystals? • role in translocation, recognition, chromatin... • PEST signals target proteins for proteosomal degradation, enriched in unstructured proteins (Singh, Proteins, 2006) • role in disease (amyloidosis) • NACP – non-ab component precursor (14 kDa), intrinsically unstructured in solution
Disorder in CREB transcriptional activator • phosphorylation modulates interaction with CBP • Wright and Dyson (JMB, 1999); Radhakrishnan (1997, 1998) • kinase-induced domain (KID) of CREB binds CBP (res 586-672) • only when phosphorylated on Ser133; forms pair of helices • disordered when de-phosphorylated (<10% a by NMR) • DH= -10.6 kcal/mol, DS = -6 kcal/mol (entropically disfavored)
Calcineurin helix must be accessible to get bound by calmodulin
enriched in P, E, K, S, and Q (charged) • depleted in W, Y, F, C, I, L, and N (hydrophobic) • low sequence complexity (Romero et al., 2001) • repeated aa’s, like collagen or silk; poly-Ala, Gly, Pro... • low entropy of aa probs (window=45) • K2<2.9: almost always disordered • different “flavors” of disorder? • clusters, Dirichlet mixtures... K2
PONDR (Obradovic) • Prediction of Naturally Disordered Regions • neural networks • input sliding window 9-21 aa; output smoothed over 9 aa • multiple classifiers (different training sets): VL1, VSL1, XL1, XC... • VL-XT (accuracy ~ 80%) • The VL-XT predictor integrates three feedforward neural networks: the VL1 predictor (Romero et al. 1997), the N-terminus predictor (XN), and the C-terminus predictor (XC) (both from Li et al. 1999). VL1 was trained using 8 long disordered regions identified from missing electron density in x-ray crystallographic studies, and 7 long disordered regions characterized by NMR. The XN and XC predictors, together called XT, were also trained using x-ray crystallographic data, where the terminal disordered regions were 5 or more amino acids in length. Coordination number is the average number of side chain neighbors that are in contact with the given side chain when it is fully buried as determined from a set of 33 non-homologous proteins.
PDB files • records: • ATOM, HETATM, TER, ENDMDL, chain ids • connectivity assumed; usually no H's • B-factors, alt conf, NMR, ligands • resolution vs. coordinate precision • poly-ala, missing, mutations, truncations, His-tag
Post-translational modifications • phosphorylation, glycosylation, lipidifaction • proteolysis • disulfide bridges • side-chain adducts: GFP, katG • covalent co-factors - PLP • oxidation of sulfhydryls • fMet - peptide deformylase; inteins • acylation, ACE? • co-factors: Fe/S proteins, hemes