Download
1 / 1
20 likes | 246 Views
Deregulated miRNAs in the most frequent RCC subtypes and UUT-UCC: Chromosomal distribution, putative target genes and molecular pathways in which they are implicated. Apostolos Zaravinos and Constantinos C Deltas.

E N D
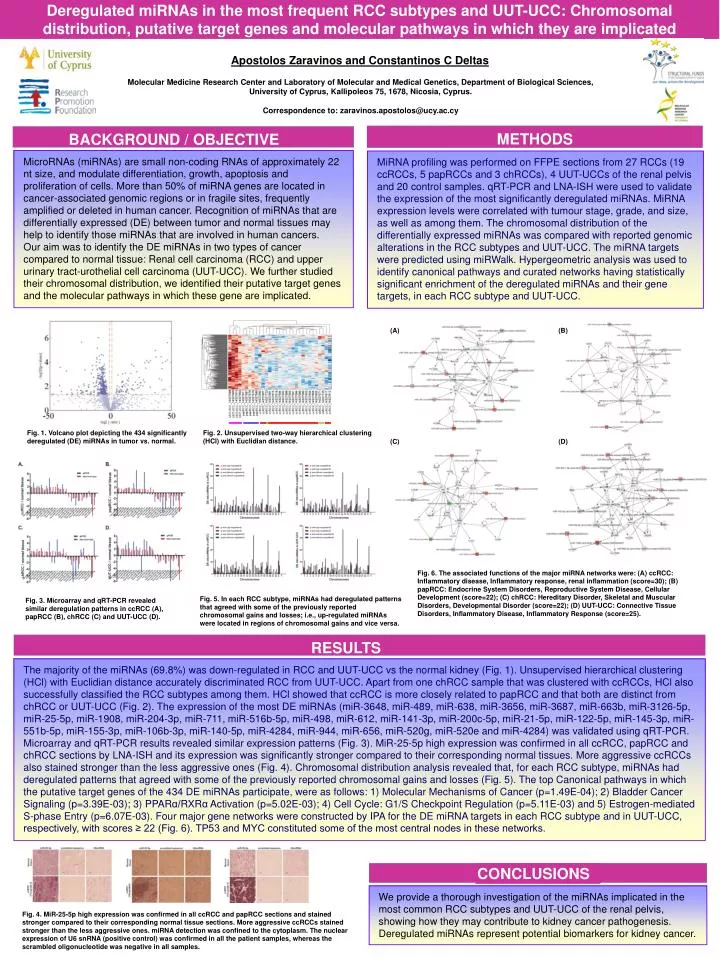
Deregulated miRNAs in the most frequent RCC subtypes and UUT-UCC: Chromosomal distribution, putative target genes and molecular pathways in which they are implicated Apostolos Zaravinos and Constantinos C Deltas Molecular Medicine Research Center and Laboratory of Molecular and Medical Genetics, Department of Biological Sciences, University of Cyprus, Kallipoleos 75, 1678, Nicosia, Cyprus. Correspondence to: zaravinos.apostolos@ucy.ac.cy MicroRNAs (miRNAs) are small non-coding RNAs of approximately 22 nt size, and modulate differentiation, growth, apoptosis and proliferation of cells. More than 50% of miRNA genes are located in cancer-associated genomic regions or in fragile sites, frequently amplified or deleted in human cancer. Recognition of miRNAs that are differentially expressed (DE) between tumor and normal tissues may help to identify those miRNAs that are involved in human cancers. Our aim was to identify the DE miRNAs in two types of cancer compared to normal tissue: Renal cell carcinoma (RCC) and upper urinary tract-urothelial cell carcinoma (UUT-UCC). We further studied their chromosomal distribution, we identified their putative target genes and the molecular pathways in which these gene are implicated. MiRNA profiling was performed on FFPE sections from 27 RCCs (19 ccRCCs, 5 papRCCs and 3 chRCCs), 4 UUT-UCCs of the renal pelvis and 20 control samples. qRT-PCR and LNA-ISH were used to validate the expression of the most significantly deregulated miRNAs. MiRNA expression levels were correlated with tumour stage, grade, and size, as well as among them. The chromosomal distribution of the differentially expressed miRNAs was compared with reported genomic alterations in the RCC subtypes and UUT-UCC. The miRNA targets were predicted using miRWalk. Hypergeometric analysis was used to identify canonical pathways and curated networks having statistically significant enrichment of the deregulated miRNAs and their gene targets, in each RCC subtype and UUT-UCC. CONCLUSIONS BACKGROUND / OBJECTIVE RESULTS METHODS (A) (B) Fig. 1. Volcano plot depicting the 434 significantly deregulated (DE) miRNAs in tumor vs. normal. Fig. 2. Unsupervised two-way hierarchical clustering (HCl) with Euclidian distance. (C) (D) Fig. 6. The associated functions of the major miRNA networks were: (A) ccRCC: Inflammatory disease, Inflammatory response, renal inflammation (score=30); (B) papRCC: Endocrine System Disorders, Reproductive System Disease, Cellular Development (score=22); (C) chRCC: Hereditary Disorder, Skeletal and Muscular Disorders, Developmental Disorder (score=22); (D) UUT-UCC: Connective Tissue Disorders, Inflammatory Disease, Inflammatory Response (score=25). Fig. 5. In each RCC subtype, miRNAs had deregulated patterns that agreed with some of the previously reported chromosomal gains and losses; i.e., up-regulated miRNAs were located in regions of chromosomal gains and vice versa. Fig. 3. Microarray and qRT-PCR revealed similar deregulation patterns in ccRCC (A), papRCC (B), chRCC (C) and UUT-UCC (D). The majority of the miRNAs (69.8%) was down-regulated in RCC and UUT-UCC vs the normal kidney (Fig. 1). Unsupervised hierarchical clustering (HCl) with Euclidian distance accurately discriminated RCC from UUT-UCC. Apart from one chRCC sample that was clustered with ccRCCs, HCl also successfully classified the RCC subtypes among them. HCl showed that ccRCC is more closely related to papRCC and that both are distinct from chRCC or UUT-UCC (Fig. 2). The expression of the most DE miRNAs (miR-3648, miR-489, miR-638, miR-3656, miR-3687, miR-663b, miR-3126-5p, miR-25-5p, miR-1908, miR-204-3p, miR-711, miR-516b-5p, miR-498, miR-612, miR-141-3p, miR-200c-5p, miR-21-5p, miR-122-5p, miR-145-3p, miR-551b-5p, miR-155-3p, miR-106b-3p, miR-140-5p, miR-4284, miR-944, miR-656, miR-520g, miR-520e and miR-4284) was validated using qRT-PCR. Microarray and qRT-PCR results revealed similar expression patterns (Fig. 3). MiR-25-5p high expression was confirmed in all ccRCC, papRCC and chRCC sections by LNA-ISH and its expression was significantly stronger compared to their corresponding normal tissues. More aggressive ccRCCs also stained stronger than the less aggressive ones (Fig. 4). Chromosomal distribution analysis revealed that, for each RCC subtype, miRNAs had deregulated patterns that agreed with some of the previously reported chromosomal gains and losses (Fig. 5). The top Canonical pathways in which the putative target genes of the 434 DE miRNAs participate, were as follows: 1) Molecular Mechanisms of Cancer (p=1.49E-04); 2) Bladder Cancer Signaling (p=3.39E-03); 3) PPARα/RXRα Activation (p=5.02E-03); 4) Cell Cycle: G1/S Checkpoint Regulation (p=5.11E-03) and 5) Estrogen-mediated S-phase Entry (p=6.07E-03). Four major gene networks were constructed by IPA for the DE miRNA targets in each RCC subtype and in UUT-UCC, respectively, with scores ≥ 22 (Fig. 6). TP53 and MYC constituted some of the most central nodes in these networks. We provide a thorough investigation of the miRNAs implicated in the most common RCC subtypes and UUT-UCC of the renal pelvis, showing how they may contribute to kidney cancer pathogenesis. Deregulated miRNAs represent potential biomarkers for kidney cancer. Fig. 4. MiR-25-5p high expression was confirmed in all ccRCC and papRCC sections and stained stronger compared to their corresponding normal tissue sections. More aggressive ccRCCs stained stronger than the less aggressive ones. miRNA detection was confined to the cytoplasm. The nuclear expression of U6 snRNA (positive control) was confirmed in all the patient samples, whereas the scrambled oligonucleotide was negative in all samples.