Download

1 / 20
200 likes | 233 Views
Explore how a probabilistic language model can enhance protein identification from tandem mass spectra, addressing current limitations and proposing a novel solution. The study compares popular approaches in computational proteomics and information retrieval fields, highlighting the significance of interdisciplinary research. Experimental results and data sets analysis provide insights into the effectiveness of the proposed method.

E N D
Protein Identification from Tandem Mass Spectra with Probabilistic Language Modeling Yiming Yang1,2, Abhay Harpale1 and Subramanian Ganaphathy1 1: Language Technologies Institute 2: Machine Learning Department School of Computer Science, Carnegie Mellon University
Outline • Motivation & Background • Two probabilistic approaches • Experiments @Yiming Yang, ECML 2009, Sept 8
Motivation • Proteins are important bio-markers for diseases, drug toxicity, therapeutic outcomes, etc. • Statistical approaches have been developed for protein identification in computational proteomics • Interdisciplinary research for comparing current solutions with successful methods in IR (information retrieval) for similar problems has been rare. • We address this research gap by • Analyzing a major limitation of popular approaches in protein ID • Proposing a new solution (Language Modeling for IR) @Yiming Yang, ECML 2009, Sept 8
The Protein ID Problem • Tandem mass (MS-MS) spectra are producedusing some chemical process on an input sample (e.g., blood) • A sample typically consists of multiple proteins. • The process segments each protein into many (hundreds) pieces, called peptides. • Peptides are further decomposed into ionized segments. • The MS-MS spectrum of a peptide is a series of spikes. • Each spike is the mass/charge (m/z) ratio of an ionized segment in the peptide. @Yiming Yang, ECML 2009, Sept 8
The Protein ID Problem (cont’d) • Protein identification requires a mapping from empirical (MS-MS) spectra to protein sequences in an DB • There are many protein sequence databases • SwissProt, for example, contains 280,000+ sequences • Each protein is defined as a sequence of amino-acid letters • Peptides in each protein are specified using cleaving rules • Each peptide has an amino-acid sequence and a corresponding theoretical (“expected”) spectrum @Yiming Yang, ECML 2009, Sept 8
Theoretical Spectra of peptides in a DB Empirical Spectra of peptides in a sample Mapping Matching • Fourier Transformation • Probabilistic Models • Heuristic Rules @Yiming Yang, ECML 2009, Sept 8
Theoretical Spectra Empirical Spectra Mapping Matching Words in L1 Words in L2 Matched Words (in L2) Doc Retrieval Matched Documents (in L2) @Yiming Yang, ECML 2009, Sept 8
Outline • Motivation & Background • Two probabilistic approaches • Experiments @Yiming Yang, ECML 2009, Sept 8
A Popular Approach in Protein ID(ProteinProphet by Nesvizhskii et al., 2003) • Given the predicted peptides based on MS-MS spectra, the probability for each candidate protein is estimated as: -- estimates the probability for a Boolean OR logic -- typically produces many false positives @Yiming Yang, ECML 2009, Sept 8
A Popular Approach in IR • Language Models (Ponte 1998; Lafferty & Zhai, 2001; …) • Query (q) is represented using a bag of words • Document (d) is represented using a bag of words • KL-divergence of the two words distributions (θqandθd ) is Cross entropy H (θq ||θd) -- not affect doc ranking -- a “soft” measure for the Boolean AND logic @Yiming Yang, ECML 2009, Sept 8
LM for Protein ID • Query language model for predicted peptides • Document language model for each protein sequence @Yiming Yang, ECML 2009, Sept 8
Outline • Motivation & Background • Two probabilistic approaches • Experiments @Yiming Yang, ECML 2009, Sept 8
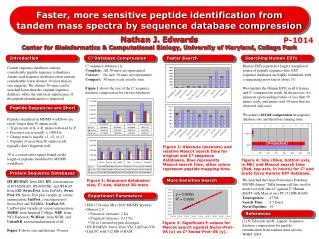
Data Sets • PPK (Purvine et al., 2003) • 2995 empirical spectra from a mixture of 35 proteins • 4535 protein sequences (325,812 unique peptides) • Mark12 • 9380 empirical spectra from a mixture of 12 proteins • 50,012 protein sequences (5,149,302 unique peptides) randomly sampled from the SwithProt database • Sigma49 • 12,498 empirical spectra from a mixture of 49 proteins • 50,049 protein sequences (2,571,642 unique peptides) randomly sampled from the SwithProt database @Yiming Yang, ECML 2009, Sept 8
Systems • Prob-AND • Our proposed method • Prob-OR • Nesvizhskii’s method, our own implementation • Conventional Vector Space Model (TFIDF-cosine) • Supported by the Lemur search engine (Callan, 2002) • X!Tandem • A popular software (online available) for protein/peptide ID • All the system, except X!Tandem, used SEQUEST to predict a set of peptides (as the “query”). • Each system produces a ranked list of proteins per query. @Yiming Yang, ECML 2009, Sept 8
Metrics • Mean Average Precision (MAP) • Standard metric in IR for evaluating ranked lists • Evaluate each ranked list from the top to each position where a true positive document is retrieved • Recall = TP/(TP + FN) • Precision = TP/(TP + FP) • TP = # of true positives, TN = # of true negatives • FP = # of false positives, FN = # of false negatives • Average the precision scores in recall intervals among 0%, 10%, 20%, …, 100% (“11-pt AVGP”) • Compute the mean of AVGP across all intervals and for all queries @Yiming Yang, ECML 2009, Sept 8
Main Results @Yiming Yang, ECML 2009, Sept 8
Statistical Significance Tests on Proportions @Yiming Yang, ECML 2009, Sept 8
Summary • The first interdisciplinary investigation/evaluation of state-of-the-art IR methods (LM and VSM) in protein identification • Prob-AND (LM) is a better choice of criterion than prob-OR in combining peptide-level evidence, improving precision significantly in the high-recall regions. • Understanding the nature of proteomic data/problems by researchers with different backgrounds (IR or ML) is hard, but, the outcome is and will be rewarding. @Yiming Yang, ECML 2009, Sept 8
Future Research • Finding the “best” protein mixture (Arnold et al., PSB 2007) • Instead of predicting each protein independently • Reduces to solving the minimum set cover problem (NP-hard) • Revised as to find the most likely protein mixture (Li et al., 2008) • Greedy approximation strategies • Using Gibbs sampling (local maxima, efficiency issues) • Better results than ProteinProphet (prob-OR) on Sigma49 • Comparative evaluation (with LM, VSM, etc.) would be informative • Scalability for high-recall predictions from very large protein databases? @Yiming Yang, ECML 2009, Sept 8
Thanks! @Yiming Yang, ECML 2009, Sept 8