Download

1 / 24
240 likes | 341 Views
Using EST Evidence to Automatically Predict Alternatively Spliced Genes. Bob Zimmermann Master’s Thesis Defense 12-12-2006. What is Genome Annotation*?. Assert the locations of protein coding genes in genomic DNA Very small part of the genome:1-2% in Hs Assert what of proteins are encoded

E N D
Using EST Evidence to Automatically Predict Alternatively Spliced Genes Bob Zimmermann Master’s Thesis Defense 12-12-2006
What is Genome Annotation*? • Assert the locations of protein coding genes in genomic DNA • Very small part of the genome:1-2% in Hs • Assert what of proteins are encoded • Give a “profile” of a genome’s products
“Gene” vs. “Transcript” • Gene - region of genomic transcription (locus) • Transcript - specific splice of a gene • Genes have one or more “isoforms” • These can lead to different protein products • Greatly influences genome diversity • 50-70% Hs genes altspliced Stated goal: find all transcripts of all genes
Way 1: Sequencing and Alignment RNA Pros: • Reliable • Inexpensive Cons: • Bias toward overexpressed genes • Bias toward short genes cDNA >gi|7732|emb|X54360.1|DMCID AGTCCACTCGTAAGAAACATAGGAATAAGACGCAGCATTCAAAAAATATTGACTTGTCTTACAAAACTGATTTTCATTGTTCGCTACTTAATATTTAGTGATA . . . genome
Way 2: Gene Prediction Pros: • No intrinsic bias • Cheap (like computers) Cons: • Won’t predict alt splices • Not extraordinarily sensitive
Way 3: Hand Annotation Pros: • Most reliable, most trusted • Most likely to pinpoint subtlety Cons: • Expensive • Won’t scale with the rate of sequencing
Can we reduce human labor? • Sequencing fl-cDNAs for all transcripts is not cost-effective • N-SCAN gets ~24k genes (most loci) in Hs • But there are at least 85k transcripts • Possible solution: ESTs--short, partial sequences of transcripts • Inexpensive • Lots of data, fast • Scales well with the rate of sequencing
Simplify: N-SCAN_EST Wei, et. al. 2006 NNNNNEEEEEIEEEEEEIIIIIIIIEEEEEIIIEEEEEENNNN
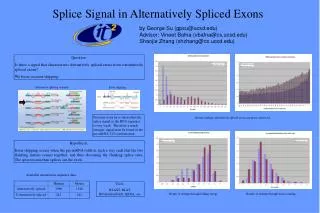
Better: Reduce Alignments Haas, et. al. 2003 PASA How can we harness this?
Method 1: MultiPass ESTSEQ • Run multiple times on same target regions …in only 1870 hours!
NEEEEIIIIOOOOIIIIIEEEE EEEMMIIIIEEEENNEEIIIIIIIIMMEEEE Method 2: AltSplice ESTSEQ
Way 3: Annotation Update • As a post-processing step, use ESTs to find alternate isoforms and correct: N-SCAN_EST prediction Haas, et. al. 2003
Fantastic • This is a very strong result but-- • A lot of this can come from fl-cDNAs alone • The amount of seq we used is costly • 255k ESTs • 21k fl-cDNAs • So where do we fit in? • With no ESTs, PASA can’t predict anything • We can
10,000 ESTs .49 Sn 100,000 ESTs .48 Sn
Conclusions • Multiple-pass prediction with different sequences predicts fancy altsplices • Heuristically reincorporating ESTs greatly improves annotations • With an order of magnitude fewer ESTs, this performs as well as N-SCAN_EST • Small sequencing projects can yield strong annotations
Caveats and Future Work • We used all annotations in Dm to train • Related species perform well • The multipass method can be efficient • Instead of naïvely running on all ESTs, only conflicting regions could be rerun
Acknowledgements • Research • Brent lab: • Sam - N-SCAN • Chaochun - N-SCAN_EST • Jeltje - ideas for MP_EST and PASA • Randy - endless parameter estimation conversations • Mani Arumugam, Beth Frazier, Aaron Tenney, Charles Comstock, Suman Kumar and Laura Langton • BDGP for vector sequences and nice ESTs • Committee: Gary Stormo, Jeremy Buhler • Love and support • Laura, my friends, my cats