Download

1 / 13

E N D
Chromosome 12 M. Pietrella1, G. Falcone1, E. Fantini1, A. Fiore1, C. Perla1, M.R. Ercolano2, A. Barone2, M.L. Chiusano2, S. Grandillo3, N. D’Agostino2, S. Melito2, S. Torre2, A. Traini2, L. Frusciante2, A. Vezzi4, S. Todesco4, M. D'Angelo4, R. Schiavon4, D. Campagna4, A. Zambon4, S. Pescarolo4, F. Levorin4, G. Valle4, G. Giuliano1 1Italian Agency for New technologies, Energy and the Environment (ENEA), Roma, Italy 2Department of Soil, Plant, Environmental and Animal Production Sciences, Univ. of Naples "Federico II", Portici, Italy 3CNR, Institute for Plant Genetics-Portici, Portici, Italy 4CRIBI Biotechnology Centre and Department of Biology, Univ. of Padova, Padova, Italy
IL mapping of BACs: workflow An optimized workflow has been developed for the mapping of BACs using esculentum/pennellii Introgression Lines
IL mapping of BACs: results The procedure has been used to confirm the map position of BACs previously mapped on Chrom 12 at Cornell and at Syngenta, or, as a service to the community, to “de novo” map novel seed BACs on the entire genome. The data are avalable on SGN.
IL mapping of “orphan” sequenced BACs Some BACs sequenced by a country have been found “a posteriori” not to map on that country’s chromosome. As a service to the community, those BACs are being assigned on a novel chromosome. The data are available on SGN.
A) B) BAC extension A tool for extending seed BACs (PABS, Todesco et al., 2008) has been developed and is available at http://tomato.cribi.unipd.it/index.html.
Genome annotation In order to contribute to the genome annotation, we developed ISOL@ (Chiusano et al, 2008), a bioinformatics resource for Solanaceae genomics. Based on EST analysis, ISOL@ allows a cross-link from the BAC sequences with other databases (transcriptome-EST and proteome-UNIPROT) to obtain a functional annotation.
Fosmid end sequencing Paired end sequences have been obtained for approx. 50,000 fosmids. Approx. 10% of the sequences match chloroplast DNA. The data are being uploaded on SGN.
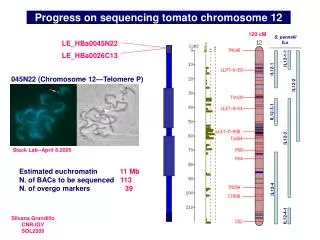
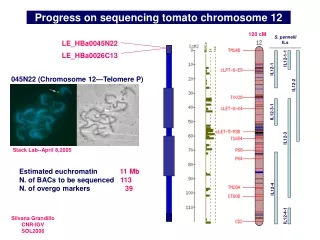
Chromosome 12 sequencing status Currently, there are 71 BACs in various steps of the sequencing process, i.e. around 62% of the projected gene-rich region. A total of 5.1 Mb non redundant sequence have been submitted (up from 1.2 Mb last year). A comparative genetic and cytogenetc map is under construction, in collaboration with H. De Jong and D. Szinay.
What does the genome look like Distribution of repeat and EST sequences in sequenced BACs Chiusano et al
454 sequencing of 24 BACs Shotgun sequencing (using MIDs) Preparation of single libraries, one per BAC, using adaptors with MIDs. There’re 12 different MIDs, therefore the 24 BACs were sorted in 2 groups: S1: BACs 1 to 12 (MIDs 1-12) S2: BACs 13 to 24 (MIDs 1-12) Sequenced in distinct regions of the PTP. Slide kindly provided by BMR-genomics, spin-off of CRIBI
454 sequencing of 24 BACs Long Paired End The same two groups of pooled BACs were hydrosheared to obtain two pools of fragments of about 3-4 kbases. The purified fragments were used to produce 2 long paired end libraries. In this case BACs were not tagged! Slide kindly provided by BMR-genomics, spin-off of CRIBI
Using the Long Paired End approach, we can obtain scaffolds of ordered and oriented contigs. ~ 3kb ~ 3kb ~ 3kb The Long Paired End Strategy After a shotgun sequencing run usually we obtain a series of contig of aligned sequences, but…. we don’t know: - how these contigs are oriented - how they are ordered - the distance between two consecutive contigs
Sequence Assembly Obtained reads were: automatically sorted in different folders according to the MID sequence (found at the beginning of each read and then trimmed). 24 folders BAC specific assembly of the shotgun reads contigs construction and, after, addition of the long paired end reads scaffolds production LPE reads (not tagged!) were assigned to the proper BAC by performing a series of BLAST against the shotgun (tagged) sequences.