Download
1 / 24
250 likes | 763 Views
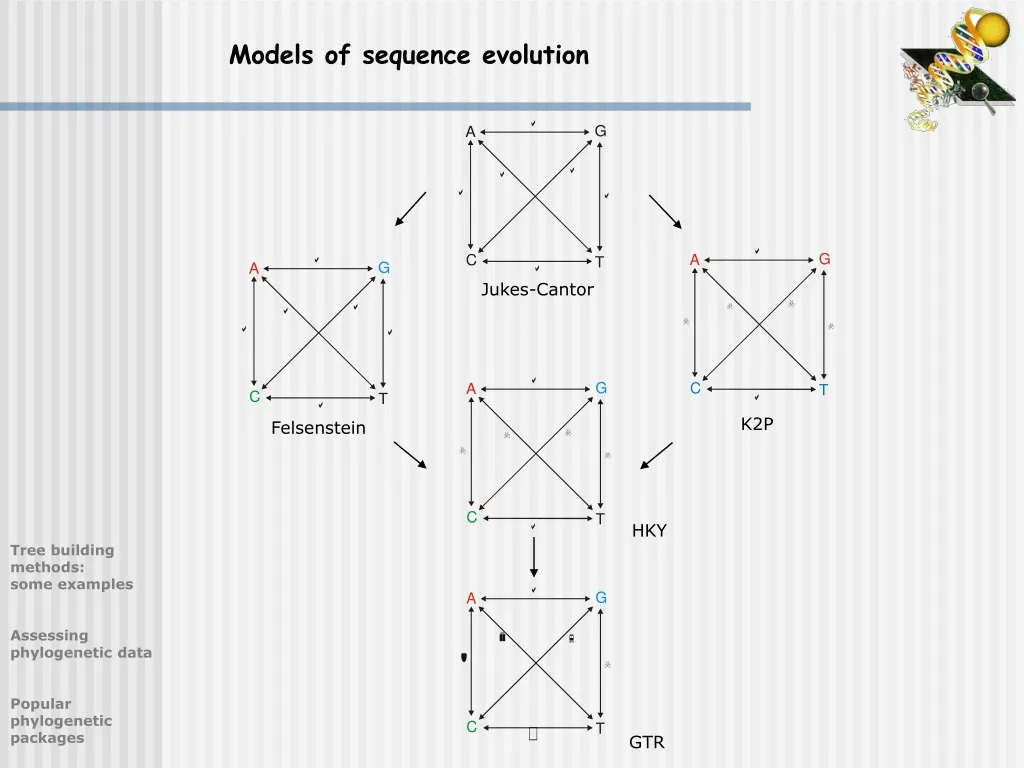
Models of sequence evolution. Jukes-Cantor. K2P. Felsenstein. HKY. Tree building methods: some examples Assessing phylogenetic data Popular phylogenetic packages. GTR. More models of sequence evolution …. Currently, there are more than 60 models described

E N D
Models of sequence evolution Jukes-Cantor K2P Felsenstein HKY Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages GTR
More models of sequence evolution … • Currently, there are more than 60 models described • plus gamma distribution and invariable sites • accuracy of models rapidly decreases for highly divergent sequences • problem: more complicated models tend to be less accurate (and slower) • How to pick an appropriate model? • use a maximum likelihood ratio test • - implemented in Modeltest 3.06 (Posada & Crandall, 1998) Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
B Equal base frequencies Null model = JC -lnL0 = 3369.2803 Alternative model = F81 -lnL1 = 3342.5513 2(lnL1-lnL0) = 53.4580 df = 3 P-value = <0.000001 C Model selected: TVM+G -lnL = 2911.3660 More models of sequence evolution … Example for Modeltest file JC = 3369.2803 F81 = 3342.5513 K80 = 3294.6611 HKY = 3124.4182 TrNef = 3114.5491 TrN = 2993.6340 K81 = 2987.6548 K81uf = 2973.5620 TIMef = 2937.6196 TIM = 2932.9878 TVMef = 2930.3450 TVM = 2922.1970 SYM = 2921.3069 GTR = 2921.1187 A Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
More models of sequence evolution … • Amino acid sequences • infinitely more complicated than nucleotide sequences • some amino acids can replace one another with relatively little effect on the structure and function of the final protein while other replacements can be functionally devastating • - from the standpoint of the genetic code, some amino acid changes can be made by a single DNA mutation while others require two or even three changes in the DNA sequence • - in practice, what has been done is to calculate tables of frequencies of all amino acid replacements within families of related protein sequences in the databanks: i.e.PAM and BLOSSUM Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
Phylogenetic Inference II Disclaimers Before describing any theoretical or practical aspects of phylogenetics, it is necessary to give some disclaimers. This area of computational biology is an intellectual minefield! Neither the theory nor the practical applications of any algorithms are universally accepted throughout the scientific community. The application of different software packages to a data set is very likely to give different answers; minor changes to a data set are also likely to profoundly change the result. Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
CS 177 Phylogenetics II Tree building methods: some examples Assessing phylogenetic data Popular phylogenetic software packages Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
helix sheet Phylogenetic Inference II Are there Correct trees?? Despite all of all problems, it is actually quite simple to use computer programs calculate phylogenetic trees for data sets Provided the data are clean, outgroups are correctly specified, appropriate algorithms are chosen, no assumptions are violated, etc., can the true, correct tree be found and proven to be scientifically valid? Unfortunately, it is impossible to ever conclusively state what is the "true" tree for a group of sequences (or a group of organisms); taxonomy is constantly under revision as new data is gathered Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
Phenetics versus cladistics • Phenetic methods construct trees (phenograms) by considering the current states of characters without regard to the evolutionary history that brought the species to their current phenotypes;phenograms are based on overall similarity • Cladistic methods construct trees (cladograms) rely on assumptions about ancestral relationships as well as on current data;cladograms are based on character evolution (e.g. shared derived characters) Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
Tree building methods Data type: genetic distance / character-state • Computational method: optimality criterion/clustering algorithmen Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
Tree building (distance based) • UPGMA • - The simplest of the distance methods is the UPGMA (Unweighted Pair Group Method using Arithmetic averages) • Many multiple alignment programs such as PILEUP use a variant of UPGMA to create a dendrogram of DNA sequences which is then used to guide the multiple alignment algorithm Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
UPGMA Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
UPGMA Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
UPGMA Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
UPGMA Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
UPGMA Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
Root UPGMA Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
Maximum Parsimony (MP) • Parsimony involves evaluating all possible trees for each vertical column of sequence character (nucleotide position) • only informative sites are considered • each tree is given a score based on the number of evolutionary changes that are needed to explain the observed data • - finally, those trees that produce the smallest number of changes (shortest trees) overall for all sequence positions are identified Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
Maximum Likelihood (ML) • Maximum Likelihood uses probability calculations based on a specific model of sequence evolution to find a tree that best accounts for the variation in a set of sequences • all possible trees for each nucleotide position are considered • the less mutations needed to fit a tree to the data, the more likely the tree • ML resembles MP in that the tree with the least number of changes will be most likely • however, ML evaluates trees using explicit evolutionary models • thus, the method can be used to explore relationships among more diverse taxa Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
Taxa (n) unrooted (2n-5)!/(2n-3(n-3)!) 2 1 3 1 4 3 5 15 6 105 7 954 8 10,395 9 135,135 10 2,027,025 . . . . . . 30 3.58 x 1036 Computational methods for finding optimal trees Possible evolutionary trees Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
Computational methods for finding optimal trees • Exact algorithms • “Guarantee” to find the optimal or “best” tree for the method of choice • Two types used in tree building: • Exhaustive search: Evaluates all possible unrooted trees, choosing the one with the best score for the method • Branch-and-bound search: Eliminates part of the tree that only contain suboptimal solutions • Heuristic algorithms • Approximate or “quick-and-dirty” methods that attempt to find the optimal tree for the method of choice, but cannot guarantee to do so • Often operate by “hill-climbing” methods Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
Search for global maximum Search for global minimum GLOBAL MAXIMUM GLOBAL MAXIMUM local maximum Rerunning heuristic searches using different input orders of taxa can help find global minima or maxima local minimum GLOBAL MINIMUM GLOBAL MINIMUM Heuristic algorithms Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages Heuristic search algorithms are input order dependent and can get stuck in local minima or maxima From NHGRI lecture, C.-B. Stewart
Assessing Phylogenetic Data Most data includes potentially misleading evidence of relationships One should not only construct phylogenetic hypotheses but should also assess what ‘confidence’ can be placed in these hypotheses Question: How much support is there for a particular clade? Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages
Assessing Phylogenetic Data How much support is there for a particular clade? Bootstrapping/Jack-knifing: Lots of randomized data sets are produced by sampling the real data with replacement (or in jackknifing, by removing some random proportion of the data); Frequencies of occurrence of groups are a measure of support for those groups Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages Problems: - Bootstrap proportions aren’t easily interpretable - no indication for how good the data are but simply for how well the tree fits the data
Popular phylogenetic software packages Review available at: http://evolution.genetics.washington.edu/phylip/software.html Tree building methods:some examples Assessing phylogenetic data Popular phylogenetic packages