Download

1 / 49
490 likes | 504 Views
Explore the concept of horizontal gene transfer (HGT) through discrepancies in species and gene classifications. Learn about new methods, applications, and future works in detecting HGT. Discover software and methods for building phylogenetic networks.

E N D
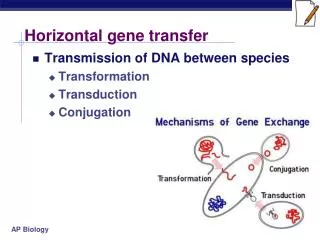
Detecting horizontal gene transfers using discrepancies in species and gene classifications Alix Boc Vladimir Makarenkov Université du Québec à Montréal
Presentation summary • Some words about phylogeny • Network models in phylogenetic analysis • What is a horizontal gene transfer (HGT)? • Description of the new method • Examples of application • Future works • T-Rex software
Recontruction of a phylogenetic tree DNA Sequences Distance Matrix Phylogenetic Tree A: CGTAAT B: CGTACG C: CGTCGA D: ACT……… E: ……………… F: ………………
Inferring phylogenetic trees • Four main approaches: • Distance-based methods • UPGMA by Michener and Sokal (1957) • ADDTREE by Sattath et Tversky (1977) • Neighbor-joining (NJ)by Saitou and Nei (1988) • UNJ and BioNJ methods by Gascuel (1997) • Fitch by Felsenstein (1997) • Weighted least-squares MW by Makarenkov and Leclerc (1999) • Maximum Parsimony (Camin and Sokal 1965; Farris 1970; Fitch 1971) • Maximum Likelihood (Felsenstein 1981) • Bayesian approach (Rannala and Yang 1996; Huelsenbeck and Ronquist 2001)
Phylogenetic mechanisms requiring a network representation • Horizontal gene transfer (i.e. lateral gene transfer) • Hybridization • Homoplasy and gene convergence • Gene duplication and gene loss
Software for building phylogenetic networks • SplitsTree, Huson (1998) • T-Rex, Makarenkov (2001) • NeighborNet, Bryant and Moulton (2002)
Methods for detecting horizontal gene transfers • Hein (1990) and Hein et al. (1995, 1996) • Haseler and Churchill (1993) • Page (1994); Page and Charleston (1998) • Charleston (1998) • Hallet and Lagergren (2001) • Mirkin, Fenner, Galperin and Koonin (2003) • V’yugin, Gelfand and Lyubetsky (2003) • Boc and Makarenkov (2003); Makarenkov, Boc and Diallo (2004)
The new model Basic ideas: Reconcile the species and gene phylogenetic trees using either a topological (Robinson and Foulds topological distance) or a metric (least-squares) criterion 2) Incorporatenecessarybiological rules into the mathematical model 3) Maintain the algorithmic time complexity polynomial
Partial gene transfer. Incorporating biological rules. Situations when a new HGT branch (a,b) can affect the evolutionary distance between species i and j, and cannot affect the distance between i1 and j.
Partial gene transfer. Incorporating biological rules (2). Three cases when the evolutionary distance between the species i and j is not affected by addition of a new HGT branch (a,b)
Partial gene transfer. Incorporating biological rules (3). No HGTs can be considered when affected branches are located on the same lineage
Partial gene transfer. Incorporating biological rules (4). No HGT can be considered when two HGTs affecting a pair of lineages intersect as shown
Partial gene transfer. Incorporating biological rules (5). • Cases (a) and (b): path between the leaves i and j is allowed to go through both HGT branches (a,b) and (a1,b1). • Cases (c) and (d) : path between the leaves i and j is not allowed to go through both HGT branches (a,b) and (a1,b1).
Sub-Tree constraint • To arrange the topological conflicts between T and T1 that are due to the • transfers between single species or their close ancestors. • To identify the transfers that have occurred deeper in the phylogeny. Timing constraint: the transfer between the branches (z,w) and (x,y) of the species tree T can be allowed if and only if the cluster regrouping both affected sub-trees is present in the gene tree T1. Here and further in the article a single branch is depicted by a plane line and a path is depicted by a wavy line.
Optimization problem : Least-squares The least-squares loss function to be minimized with an unknown length l of the HGT branch (a,b): Q(ab,l) = + min d(i,j) - the minimum path-length distance between the leaves (i.e. taxa) i and j in the tree T (i,j) - the given dissimilarity value between i and j dist(i,j) = d(i,j) – Min { d(i,a) + d(j,b); d(j,a) + d(i,b) }
Optimization problem : Robinson and Foulds topological distance The topological distance of Robinson and Foulds (1981) between two phylogenetic trees is equal to the minimum number of elementary operations consisting of merging or splitting vertices necessary to transform one tree into another.
Robinson and Foulds topological distance Robinson and Foulds distance between T and T1 is 2. The HGT minimizing the Robinson and Foulds topological distance between the species and gene phylogenetic trees can be considered as the best candidate to reconcile the species and gene phylogenies.
Input file for our program Set X of Taxa = {A,B,C,D,E,F} 6 A 0 2 3 5 5 4 B 2 0 3 5 5 4 C 3 3 0 4 4 3 D 5 5 4 0 2 3 E 5 5 4 2 0 3 F 4 4 3 3 3 0 A 0 4 4 2 4 4 B 4 0 4 4 2 4 C 4 4 0 4 4 2 D 2 4 4 0 4 4 E 4 2 4 4 0 4 F 4 4 2 4 4 0 6 Distance Matrix for the species tree Distance Matrix for the gene tree
Program options • Optimization criterion : Least-Squares or Robinson and Foulds distance. • Type of scenario : Unique or Multiple. • Maximum number of HGTs. • Position of the root.
Algorithm : unique scenario Begin Reconstruction of the species tree T Reestimate the length of each branch in T While Optimization criterion > 0 loop Test all possible HGTs Add the best HGT Reestimate the length of each branch in T Compute the value of the optimization criterion EndLoop End
Algorithm : multiple scenario Begin Reconstruction of the species tree T Reestimate the length of each branch in T Test all connections between pairs of branches Establish a list of HGTs ordered according to the optimization criterion. End
Algorithm : Step 1 • Reconstruction of the species tree T with Neighbor Joinning • Set X of n taxa • Binary tree: internal nodes are all of degree 3, 2n-3 branches • T is explicitly rooted
Algorithm : Step 2 • Comparing the gene tree T1 and the species tree T Criterion 2 : Reestimate the length of each branch of the species tree T according to the distances in T1. LS - Least-Squares coefficient between distances in T and T1 If LS == 0 then There is no HGTs Else Step 3 (next slide) End if Criterion 1 : RF - Robinson and Foulds distance between T and T1 If RF == 0 then There is no HGTs Else Step 3 (next slide) End if
Algorithm : Step 3 • Multiple Scenario • Test all connections between pairs of branches. • Reestimate the length of each branch in T according to the gene distance matrix. • Establish a list of HGTs ordered according • to the least-squares coefficient or the • Robinson-Foulds distance.
Algorithm : Step 3 Species Tree Upcoming HGT1 Species Tree + HGT1 Upcoming HGT2 Species Tree + HGT2 Upcoming HGT3 Species Tree + HGT3 (Gene Tree) • Unique Scenario • The best HGT found is added to the species tree. • The length of each branch is reestimated according to the gene tree. • RF distance or LS coefficient are computed. 1 2 3
output Type de scenario : Unique Liste des aretes et leur longueur de l'arbre d'especes construit avec NJ 1 7---B 1.800000 2 8---C 1.800000 3 9---D 1.800000 4 10---9 0.000020 5 9---E 1.800000 6 10---F 1.800000 7 7---A 1.800000 8 7---8 0.000020 9 10---8 0.000020 Le critere des moindres carres LS pour l'arbre d'especes dont les branches sont evaluees en fonction de l'arbre de gene est: 9.600160 La racine se trouve sur la branche 8--10 ===================== TLG #1 ====================== Menant de la branche 7--B a la branche 10--9 LS = 5.333387 RF = 4 ===================== TLG #2 ====================== Menant de la branche A--7 a la branche 9--D LS = 0.000000 RF = 0
Application example 1 Horizontal transfer of the Rubisco Large subunit gene Delwiche, C.F., and J. D. Palmer. 1996. Rampant Horizontal Transfer and Duplication of Rubisco Genes in Eubacteria and Plastids. Mol. Biol. Evol. 13:873-882.
Delwiche and Palmer (1996) - hypotheses of HGTs 1- Cyanobacteria → γ-Proteobacteria 2- α-Proteobacteria → Red and brown algae 3- γ-Proteobacteria →α-Proteobacteria 4- γ-Proteobacteria →β-Proteobacteria
HGTs of the rbcL gene 1 4 3 6 7 2 5 8
HGTs of the rbcL gene - comparison Hypotheses by Delwiche and Palmer (1996) 1- Cyanobacteria → γ-Proteobacteria 2- α-Proteobacteria → Red and brown algae 3- γ-Proteobacteria →α-Proteobacteria 4- γ-Proteobacteria →β-Proteobacteria Solution 1. a-Proteobacteria →β-Proteobacteria 2. α-Proteobacteria → Red and brown algae 3. b-Proteobacteria →γ-Proteobacteria 4. b-Proteobacteria →a-Proteobacteria 5. γ-Proteobacteria →Cyanobacteria 6. β-Proteobacteria →γ-Proteobacteria 7. γ-Proteobacteria →β-Proteobacteria 8. Cyanobacteria →γ-Proteobacteria
Application example 2 Horizontal transfers of the protein rpl12e Data taken from: Matte-Tailliez O., Brochier C., Forterre P. & Philippe H. Archaeal phylogeny based on ribosomal proteins. (2002). Mol. Biol. Evol. 19, 631-639.
Rpl12e HGTs Assumed HGTs of the rpl12e gene involved the clusters of Crenarchaeota and Thermoplasmatales (Matte-Tailliez, 2004) Species tree Rpl12e gene tree
Reconciliation scenario 74% 3 60% 2 69% 4 60% 5 1 55%
Application example 3 • Horizontal transfers of the PheRS synthetase • Data taken from: • Woese, C. R., G. Olsen, M. Ibba, and D. Söll. 2000. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 64:202-236.
Reconciliation scenario 60% 4 85% 2 5 65% 88% 1 62% 3
T-REX — Tree and Reticulogram Reconstruction1 Downloadable from http://www.info.uqam.ca/~makarenv/trex.html Authors: Vladimir Makarenkov Versions: Windows 9x/NT/2000/XP and Macintosh With contributions from A. Boc, P. Casgrain, A. B. Diallo, O. Gascuel, A. Guénoche, P.-A. Landry, F.-J. Lapointe, B. Leclerc, and P. Legendre. ________ 1Makarenkov, V. 2001. T-REX: reconstructing and visualizing phylogenetic trees and reticulation networks. Bioinformatics 17: 664-668.
T-Rex : Multiple scenario screenshot Bioinformatics software
Future developments • Maximum Likelihood model • Maximum Parsimony model • Decreasing the running time
Bibliography • Boc, A. and Makarenkov, V. (2003), New Efficient Algorithm for Detection of Horizontal Gene Transfer Events, Algorithms in Bioinformatics, G. Benson and R. Page (Eds.), 3rd Workshop on Algorithms in Bioinformatics, Springer-Verlag, pp. 190-201. • Delwiche, C.F., and J. D. Palmer (1996). Rampant Horizontal Transfer and Duplication of Rubisco Genes in Eubacteria and Plastids. Mol. Biol. Evol. 13:873-882. • Makarenkov,V. (2001), T-Rex: reconstructing and visualizing phylogenetic trees and reticulation networks. Bioinformatics, 17, 664-668. • Makarenkov, V., Boc, A., Delwiche, C.F. and Philippe, H. (2005), A novel approach for detecting horizontal gene transfers: Modeling partial and complete gene transfer scenarios, submittedMol. Biol. Evol. • Makarenkov, V., Boc, A. and Diallo A.B. (2004), Representing Lateral gene transfer in species classification. Unique scenario, IFCS’2004 proceedings, Chicago. • Matte-Tailliez O., Brochier C., Forterre P. & Philippe H. (2002). Archaeal phylogeny based on ribosomal proteins. Mol. Biol. Evol. 19, 631-639. • Robinson, D.R. and Foulds L.R. (1981), Comparison of phylogenetic trees, Mathematical Biosciences 53, 131-147. • Woese, C. R., G. Olsen, M. Ibba, and D. Söll. 2000. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol. Mol. Biol. Rev. 64:202-236.