Download
1 / 15
150 likes | 319 Views
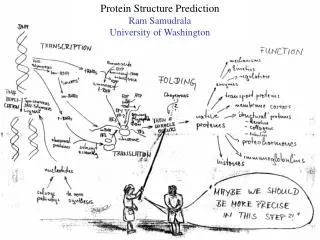
Modelling Genome Structure and Function Ram Samudrala University of Washington. Rationale for understanding protein structure and function. structure determination structure prediction. Protein structure - three dimensional - complicated - mediates function. homology

E N D
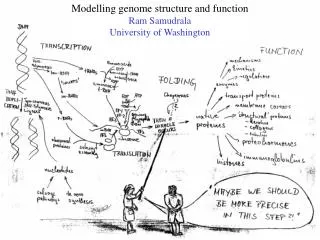
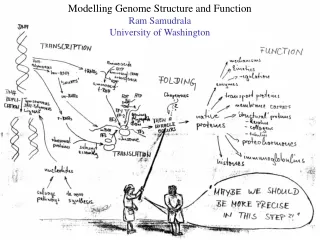
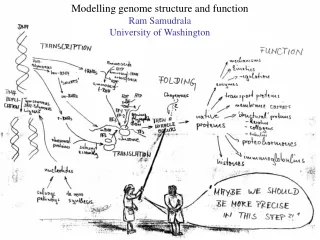
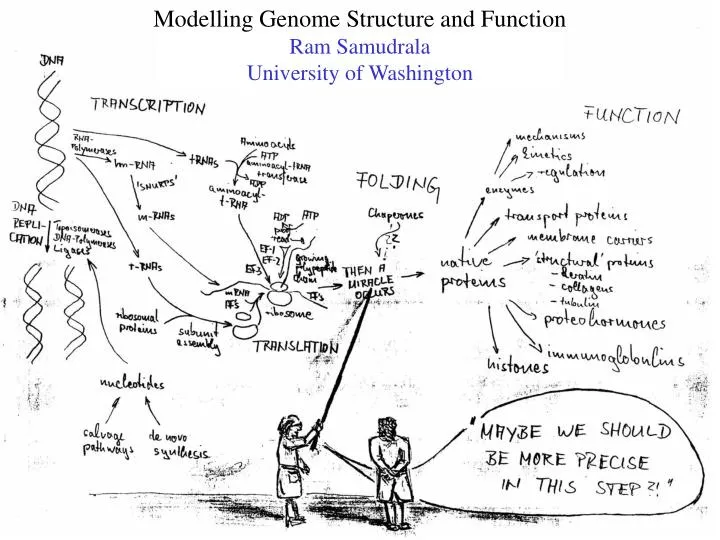
Modelling Genome Structure and Function Ram Samudrala University of Washington
Rationale for understanding protein structure and function structure determination structure prediction Protein structure - three dimensional - complicated - mediates function homology rational mutagenesis biochemical analysis model studies Protein sequence -large numbers of sequences, including whole genomes ? Protein function - rational drug design and treatment of disease - protein and genetic engineering - build networks to model cellular pathways - study organismal function and evolution
Comparative modelling of protein structure scan align KDHPFGFAVPTKNPDGTMNLMNWECAIP KDPPAGIGAPQDN----QNIMLWNAVIP ** * * * * * * * ** … … build initial model construct non-conserved side chains and main chains minimum perturbation graph theory, semfold refine physical functions de novo simulation
Comparative modelling at CASP alignment side chain short loops longer loops BC excellent ~ 80% 1.0 Å 2.0 Å CASP1 poor ~ 50% ~ 3.0 Å > 5.0 Å CASP2 fair ~ 75% ~ 1.0 Å ~ 3.0 Å CASP3 fair ~75% ~ 1.0 Å ~ 2.5 Å CASP4 fair ~75% ~ 1.0 Å ~ 2.0 Å CASP4: overall model accuracy ranging from 1 Å to 6 Å for 50-10% sequence identity **T111/eno – 1.7 Å (430 residues; 51%) **T122/trpa – 2.9 Å (241 residues; 33%) **T128/sodm – 1.0 Å (198 residues; 50%) **T125/sp18 – 4.4 Å (137 residues; 24%) **T112/dhso – 4.9 Å (348 residues; 24%) **T92/yeco – 5.6 Å (104 residues; 12%)
Ab initio prediction of protein structure select sample conformational space such that native-like conformations are found hard to design functions that are not fooled by non-native conformations (“decoys”) astronomically large number of conformations 5 states/100 residues = 5100 = 1070
Semi-exhaustive segment-based folding fragments from database 14-state f,y model generate … … monte carlo with simulated annealing conformational space annealing, GA minimise … … all-atom pairwise interactions, bad contacts compactness, secondary structure filter EFDVILKAAGANKVAVIKAVRGATGLGLKEAKDLVESAPAALKEGVSKDDAEALKKALEEAGAEVEVK
Ab initio prediction at CASP Before CASP (BC): “solved” (biased results) CASP2: worse than random with one exception CASP1: worse than random CASP3: consistently predicted correct topology - ~ 6.0 Å for 60+ residues CASP4: consistently predicted correct topology - ~4-6.0 A for 60-80+ residues *T98/sp0a – 6.0 Å (60 residues; 37-105) **T102/as48 – 5.3 Å (70 residues; 1-70) **T97/er29 – 6.0 Å (80 residues; 18-97) **T106/sfrp3 – 6.2 Å (70 residues; 6-75) **T110/rbfa – 4.0 Å (80 residues; 1-80) *T114/afp1 – 6.5 Å (45 residues; 36-80)
Computational aspects of structural genomics A. sequence space B. comparative modelling C. fold recognition * * * * E. target selection F. analysis D. ab initio prediction * * * * * * * * * * * * * * * * * * * * targets (Figure idea by Steve Brenner.)
Computational aspects of functional genomics structure based methods microenvironment analysis structure comparison sequence based methods sequence comparison motif searches phylogenetic profiles domain fusion analyses zinc binding site? homology function? + assign function to entire protein space * G. assign function * * * * * + experimental data
Modelling structure and function of the Oryza sativa (rice) genome 6813 coding sequences 3149 without a product annotation 816 classified as hypothetical protein 1187 with a hypothetical function ~30 % with known homologs in PDB Most common functions (from PROSITE) ATP/GTP-binding site motif A (P loop) Serine/Threonine protein kinase active site EF-hand (Calcium binding) Cytochrome C Heme binding site Most common functions (from annotations) Reverse transcriptase Nucleotide Binding Site (NBS) Serine/Threonine protein kinase Chitinase
Bioverse webserver sequence summary structure summary function summary http://bioverse.compbio.washington.edu see another variant open/close subgroup list links (or follow) mapping to sequence
Bioverse webserver sequence summary sequence evidence for sequence structure summary secondary structure tertiary structure evidence for tertiary structure structural similarity to another protein structural similarity to another protein evidence for similarity structural similarity to another protein
Bioverse webserver sequence summary structure summary function summary function 1 function 2 evidence for function 2 functional similarity to another protein functional similarity to another protein evidence for similarity functional similarity to another protein
Take home message Acknowledgements Jason McDermott Yi-Ling Chen Levitt and Moult groups Prediction of protein structure and function can be used to model whole genomes to understand organismal function and evolution