Download
1 / 62
630 likes | 798 Views
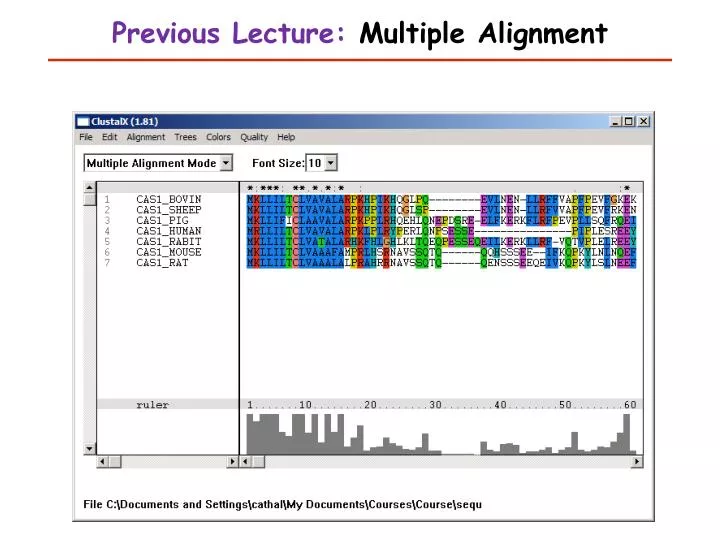
Previous Lecture: Multiple Alignment. This Lecture. Introduction to Biostatistics and Bioinformatics Motifs. Learning Objectives. Restriction sites Finding genes in DNA sequences Regulatory sites in DNA Protein signals (transport and processing)

E N D
This Lecture Introduction to Biostatistics and Bioinformatics Motifs
Learning Objectives • Restriction sites • Finding genes in DNA sequences • Regulatory sites in DNA • Protein signals (transport and processing) • Protein functional domains & motif databases • Regular Expressions • Position Specific Scoring Matrix & Hidden Markov Models
Restriction Sites • Bacteria make restriction enzymes that cut DNA at specific sequences (4-8 base patterns) • Very simple to find these patterns - can even use the “Find” function of your web browser or word processor • Open any page of text and look for “CAT” • you now have a restriction site search program!
NEBcutter2 http://tools.neb.com/NEBcutter2/
Translate (in all 6 reading frames) and look for similarity to known protein sequences Look for long Open Reading Frames (ORFs) between start and stop codons (start=ATG, stop=TAA, TAG, TGA) Look for known gene markers TAATAA box, intron splice sites, etc. Statistical methods (codon preference) Finding Genes in Genomic DNA
GCCACATGTAGATAATTGAAACTGGATCCTCATCCCTCGCCTTGTACAAAAATCAACTCCAGATGGATCTAAGATTTAAATCTAACACCTGAAACCATAAAAATTCTAGGAGATAACACTGGCAAAGCTATTCTAGACATTGGCTTAGGCAAAGAGTTCGTGACCAAGAACCCAAAAGCAAATGCAACAAAAACAAAAATAAATAGGTGGGACCTGATTAAACTGAAAAGCCTCTGCACAGCAAAAGAAATAATCAGCAGAGTAAACAGACAACCCACAGAATGAGAGAAAATATTTGCAAACCATGCATCTGATGACAAAGGACTAATATCCAGAATCTACAAGGAACTCAAACAAATCAGCAAGAAAAAAATAACCCCATCAAAAAGTGGGCAAAGGAATGAATAGACAATTCTCAAAATATACAAATGGCCAATAAACATACGAAAAACTGTTCAACATCACTAATTATCAGGGAAATGCAAATTAAAACCACAATGAGATGCCACCTTACTCCTGCAAGAATGGCCATAATAAAAAAAAATCAAAAAAGAATAAATGTTGGTGTGAATGTGGTGAAAAGAGAACACTTTGACACTGCTGGTGGGAATGGAAACTAGTACAACCACTGTGGAAAACAGTACCGAGATTTCTTAAAGAACTACAAGTAGAACTACCATTTGATCCAGCAATCCCACTACTGGGTATCTACCCAGAGGAAAAGAAGTCATTATTTGAAAAAGACACTTGTACATACATGTTTATAGCAGCACAATTTGCAATTGCAAAGATATGGAACCAGTCTAAATGCCCATCAACCAACAAATGGATAAAGAAAATATGGTATATATACACCATGGAACACTACTCAGCCATAAAAAGGAACAAAATAATGGCAACTCACAGATGGAGTTGGAGACCACTATTCTAAGTGAAATAACTCAGGAATGGAAAACCAAATATTGTATGTTCTCACTTATAAGTGGGAGCTAAGCTATGAGGACAAAAGGCATAAGAATTATACTATGGACTTTGGGGACTCGGGGGAAAGGGTGGGAGGGGGATGAGGGACAAAAGACTACACATTGGGTGCAGTGTACACTGCTGAGGTGATGGGTGCACCAAAATCTCAGAAATTACCACTAAAGAACTTATCCATGTAACTAAAAACCACCTCTACCCAAATAATTTTGAAATAAAAAATAAAAATATTTTAAAAAGAACTCTTTAAAATAAATAATGAAAAGCACCAACAGACTTATGAACAGGCAATAGAAAAAATGAGAAATAGAAAGGAATACAAATAAAAGTACAGAAAAAAAATATGGCAAGTTATTCAACCAAACTGGTAATTTGAAATCCAGATTGAAATAATGCAAAAAAAAGGCAATTTCTGGCACCATGGCAGACCAGGTACCTGGATGATCTGTTGCTGAAAACAACTGAAAATGCTGGTTAAAATATATTAACACATTCTTGAATACAGTCATGGCCAAAGGAAGTCACATGACTAAGCCCACAGTCAAGGAGTGAGAAAGTATTCTCTACCTACCATGAGGCCAGGGCAAGGGTGTGCACTTTTTTTTTTCTTCTGTTCATTGAATACAGTCACTGTGTATTTTACATACTTTCATTTAGTCTTATGACAATCCTATGAAACAAGTACTTTTAAAAAAATTGAGATAACAGTTGCATACCGTGAAATTCATCCATTTAAAGTGAGCAATTCACAGGTGCAGCTAGCTCAGTCAGCAGAGCATAAGACTCTTAAAGTGAACAATTCAGTGCTTTTTAGTATATTCACAGAGTTGTGCAACCATCACCACTATCTAATTGGTCTTAGTCTGTTTGGGCTGCCATAACAAAATACCACAAACTGGATAGCTCATAAACAACAGGCATTTATTGCTCACAGTTCTAGAGGCTGGAAGTGCAAGATTAAGATGCCAGCAGATTCTGTGTCTGCTGAGGGCCTGTTCCTCATAGAAGGTGCCCTCTTGCTGAATTCTCACATGGTGGAAGGGGGAAAACAAGCTTGCATTGCAAAGAGGTGGGCCTCTTTAATCCCAAAGGCCCCACCTCTAAAAGGCCCCACTTCTGAATACCATTACATTGAGAATTAAGTTTCAACATAGGAATTTGGGGGAACACAAATATCCAGACTGTAGCATAATTCCAGAACGGATTCATGCCACATGTAGATAATTGAAACTGGATCCTCATCCCTCGCCTTGTACAAAAATCAACTCCAGATGGATCTAAGATTTAAATCTAACACCTGAAACCATAAAAATTCTAGGAGATAACACTGGCAAAGCTATTCTAGACATTGGCTTAGGCAAAGAGTTCGTGACCAAGAACCCAAAAGCAAATGCAACAAAAACAAAAATAAATAGGTGGGACCTGATTAAACTGAAAAGCCTCTGCACAGCAAAAGAAATAATCAGCAGAGTAAACAGACAACCCACAGAATGAGAGAAAATATTTGCAAACCATGCATCTGATGACAAAGGACTAATATCCAGAATCTACAAGGAACTCAAACAAATCAGCAAGAAAAAAATAACCCCATCAAAAAGTGGGCAAAGGAATGAATAGACAATTCTCAAAATATACAAATGGCCAATAAACATACGAAAAACTGTTCAACATCACTAATTATCAGGGAAATGCAAATTAAAACCACAATGAGATGCCACCTTACTCCTGCAAGAATGGCCATAATAAAAAAAAATCAAAAAAGAATAAATGTTGGTGTGAATGTGGTGAAAAGAGAACACTTTGACACTGCTGGTGGGAATGGAAACTAGTACAACCACTGTGGAAAACAGTACCGAGATTTCTTAAAGAACTACAAGTAGAACTACCATTTGATCCAGCAATCCCACTACTGGGTATCTACCCAGAGGAAAAGAAGTCATTATTTGAAAAAGACACTTGTACATACATGTTTATAGCAGCACAATTTGCAATTGCAAAGATATGGAACCAGTCTAAATGCCCATCAACCAACAAATGGATAAAGAAAATATGGTATATATACACCATGGAACACTACTCAGCCATAAAAAGGAACAAAATAATGGCAACTCACAGATGGAGTTGGAGACCACTATTCTAAGTGAAATAACTCAGGAATGGAAAACCAAATATTGTATGTTCTCACTTATAAGTGGGAGCTAAGCTATGAGGACAAAAGGCATAAGAATTATACTATGGACTTTGGGGACTCGGGGGAAAGGGTGGGAGGGGGATGAGGGACAAAAGACTACACATTGGGTGCAGTGTACACTGCTGAGGTGATGGGTGCACCAAAATCTCAGAAATTACCACTAAAGAACTTATCCATGTAACTAAAAACCACCTCTACCCAAATAATTTTGAAATAAAAAATAAAAATATTTTAAAAAGAACTCTTTAAAATAAATAATGAAAAGCACCAACAGACTTATGAACAGGCAATAGAAAAAATGAGAAATAGAAAGGAATACAAATAAAAGTACAGAAAAAAAATATGGCAAGTTATTCAACCAAACTGGTAATTTGAAATCCAGATTGAAATAATGCAAAAAAAAGGCAATTTCTGGCACCATGGCAGACCAGGTACCTGGATGATCTGTTGCTGAAAACAACTGAAAATGCTGGTTAAAATATATTAACACATTCTTGAATACAGTCATGGCCAAAGGAAGTCACATGACTAAGCCCACAGTCAAGGAGTGAGAAAGTATTCTCTACCTACCATGAGGCCAGGGCAAGGGTGTGCACTTTTTTTTTTCTTCTGTTCATTGAATACAGTCACTGTGTATTTTACATACTTTCATTTAGTCTTATGACAATCCTATGAAACAAGTACTTTTAAAAAAATTGAGATAACAGTTGCATACCGTGAAATTCATCCATTTAAAGTGAGCAATTCACAGGTGCAGCTAGCTCAGTCAGCAGAGCATAAGACTCTTAAAGTGAACAATTCAGTGCTTTTTAGTATATTCACAGAGTTGTGCAACCATCACCACTATCTAATTGGTCTTAGTCTGTTTGGGCTGCCATAACAAAATACCACAAACTGGATAGCTCATAAACAACAGGCATTTATTGCTCACAGTTCTAGAGGCTGGAAGTGCAAGATTAAGATGCCAGCAGATTCTGTGTCTGCTGAGGGCCTGTTCCTCATAGAAGGTGCCCTCTTGCTGAATTCTCACATGGTGGAAGGGGGAAAACAAGCTTGCATTGCAAAGAGGTGGGCCTCTTTAATCCCAAAGGCCCCACCTCTAAAAGGCCCCACTTCTGAATACCATTACATTGAGAATTAAGTTTCAACATAGGAATTTGGGGGAACACAAATATCCAGACTGTAGCATAATTCCAGAACGGATTCAT
Intron/Exon structure • Gene finding programs work well in bacteria • None of the gene prediction programs do a very good job of predicting eukaryotic intron/exon boundaries • The only reasonable gene models are based on alignment of cDNAs to genome sequence • >50% of all human genes still do not have an accurate coding sequence defined (transcription start, intron splice sites)
GRAIL: Oak Ridge Natl. Lab, Oak Ridge, TN http://compbio.ornl.gov/grailexp ORFfinder: NCBI http://www.ncbi.nlm.nih.gov/gorf/gorf.html DNA translation:Univ. of Minnesota Med. School http://alces.med.umn.edu/webtrans.html GenLang http://cbil.humgen.upenn.edu/~sdong/genlang.html BCM GeneFinder:Baylor College of Medicine, Houston, TX http://dot.imgen.bcm.tmc.edu:9331/seq-search/gene-search.html http://dot.imgen.bcm.tmc.edu:9331/gene-finder/gf.html Gene Finding on the Web
Truth? • There may not be a "correct" answer to the gene finding problem • Some genes have more than one start and stop position on the DNA • Alternative splicing (a portion of the DNA is sometimes in an exon, sometimes in an intron) • Pseudogenes - look like genes, but no longer function • All computational gene predictions need to be experimentally verified (RNA-seq!!)
Genomic Sequence • Once each gene is located on the chromosome, it becomes possible to get upstream genomic sequence • This is where transcription factor (TF) binding sites are located • promoters and enhancers • Search for known TF sites, and discover new ones (among co-regulated genes)
Phage CRO repressor bound to DNA Andrew Coulson & Roger Sayles with RasMol, Univ. of Edinburgh 1993
JASPAR: a curated, non-redundant set of transcription factor binding sites from published articles (currently 593 non-redundant matrics). UniProbe:binding sites of transcription factors determined by in vitro protein binding microarray(data for 406 DNA binding proteins on all k-mers) TransFac Became a private for profit company (BIOBASE/Quiagen) Stopped adding new entries to public data in 2005 The Eukaryotic Promoter Database (EPD) 1314 entries taken directly from scientific literature Many DNA Regulatory Sequences are Known
Position Scoring Matrix Biopython Bio.motifs package (similar to BioPerl TFBS) Count matrix: 0 1 2 3 4 5 A: 4.00 19.00 0.00 0.00 0.00 0.00 C: 16.00 0.00 20.00 0.00 0.00 0.00 G: 0.00 1.00 0.00 20.00 0.00 20.00 T: 0.00 0.00 0.00 0.00 20.00 0.00 Normalized position weight matrix (with pseudocounts) = probability of each base Position Specific Scoring Matrix (log odds ratios of matrix vs background): 0 1 2 3 4 5 A: 0.22 0.69 0.09 0.09 0.09 0.09 C: 0.59 0.09 0.72 0.09 0.09 0.09 G: 0.09 0.12 0.09 0.72 0.09 0.72 T: 0.09 0.09 0.09 0.09 0.72 0.09 0 1 2 3 4 5 A: -0.19 1.46 -1.42 -1.42 -1.42 -1.42 C: 1.25 -1.42 1.52 -1.42 -1.42 -1.42 G: -1.42 -1.00 -1.42 1.52 -1.42 1.52 T: -1.42 -1.42 -1.42 -1.42 1.52 -1.42 Positive scores show that a base is more likely to come from the motif, negative scores are more likely to come from background >>> m.consensus Seq('CACGTG', IUPACUnambiguousDNA()) >>>m.weblogo("mymotif.png")
Motif Search Methods Exact Match >>> match = seq.count('CACGTG') Regular Expression Match >>> match = re.search(r'[CA][AG]CG[TC]G', seq) PSSM Search >>> from Bio import motifs >>> for position, score in pssm.search(seq, threshold=7.0): ... print("Position %d: score = %5.3f" % (position, score)) ... Position 0: score = 5.622 Position -20: score = 4.601 Position 10: score = 3.037 Position 13: score = 5.738 Threshold of log-odds 7 = 100x more likely to occur in motif than random backgroundNegative positions are on - strand A highly selective motif should only match once (or zero times) in each sequence tested.
DE IFI-6-16 (interferon-induced gene 6-16); G000176. SQ gGGAAAaTGAAACT SF -127 ST -89 BFT00428 ISGF-3; Quality: 6; Species: human, Homo sapiens. • Most TF binding sites are determined by just a few base pairs (typically 6-12) • Sequence is variable (consensus) • This is not enough information for proteins to locate unique promoters for each gene in a 3 billion base genome • TF's bind cooperatively and combinatorially • The key is in the location in relation to each other and to the transcription units of genes + epigenetic factors • Can use phylogenetic conservation to help predict binding sites TF Binding sites lack information
Web tools for TFBS Promoter Scan: NIH Bioinformatics (BIMAS) http://www-bimas.cit.nih.gov/molbio/proscan/ Signal Scan: NIH Bioinformatics (BIMAS) – uses old TransFac database http://www-bimas.cit.nih.gov/molbio/signal/ TFSEARCH (uses 1998 version of TransFac)http://www.cbrc.jp/research/db/TFSEARCH.html JASPAR (search motifs in one sequence), ConSitehttp://jaspar.genereg.net/http://consite.genereg.net/ Toucan workbench for regulatory sequence analysis https://gbiomed.kuleuven.be/english/research/50000622/lcb/tools/toucan TargetFinder: Telethon Inst.of Genetics and Medicine, Milan, Italy http://www.targetfinder.org/index.php/findtargets RSAT: Regulatory Sequence Analysis Toolkithttp://rsat.ulb.ac.be/rsat/ MotifMogul: A web server that enables the analysis of multiple DNA sequences with PWM from JASPAR and TRANSFAC using 3 different algorithms (CLOVER, MotifLocator, MotifScanner)http://xerad.systemsbiology.net/MotifMogulServer/index.html
Molecular properties (pH, mol. wt. isoelectric point, hydrophobicity) Motifs (signal peptide, coiled-coil, trans-membrane, etc.) Protein Families Secondary Structure (helix vs. beta-sheet) 3-D prediction, Threading Protein Sequence Analysis
Proteins are linear polymers of 20 amino acids Chemical properties of the protein are determined by its amino acids Molecular wt., pH, isoelectric point are simple calculations from amino acid composition Hydrophobicity is a property of groups of amino acids - best examined as a graph Chemical Properties of Proteins
Hydrophobicity Plot P53_HUMAN (P04637) human cellular tumor antigen p53 Kyte-Doolittle hydrophilicty, window=19
Web Sites for Simple Protein Analysis • Protein Hydrophobicity Server: Bioinformatics Unit, Weizmann Institute of Science , Israel http://bioinformatics.weizmann.ac.il/hydroph/ • SAPS - statistical analysis of protein sequences: composition, charge, hydrophobic and transmembrane segments, cysteine spacings, repeats and periodicity http://www.isrec.isb-sib.ch/software/SAPS_form.html
EMBOSS Protein Analysis Toolkit • plotorf:simple open reading frame finder • Garnier: predicts 2ndary structure • Charge: plot of protein charge • Octanol: hydrophobicity plot • Pepwindow: hydropathy plot • pepinfo:plotsprotein secondary structure and hydrophobicity in parallel panels • tmap: predict transmembrane regions • Topo: draws a map of transmembrane protein • Pepwheel: shows protein sequence as helical wheel • Pepcoil: predicts coiled-coil domains • Helixturnhelix: predicts helix-turn-helix domains
Common structural motifs Membrane spanning Signal peptide Coiled coil Helix-turn-helix Simple Motifs
Protein Signal Peptides • Proteins are sorted within the cell using 20-25 amino acid tags at their 5' end (beginning) • Chopped off once they reach their destination
Protein Signal Prediction • ChloroP - Prediction of chloroplast transit peptides • LipoP - Prediction of lipoproteins and signal peptides in Gram negative bacteria • MITOPROT - Prediction of mitochondrial targeting sequences • PATS - Prediction of apicoplast targeted sequences • PlasMit - Prediction of mitochondrial transit peptides in Plasmodium falciparum • Predotar - Prediction of mitochondrial and plastid targeting sequences • PTS1 - Prediction of peroxisomal targeting signal 1 containing proteins • SignalP - Prediction of signal peptide cleavage sites・
Common structural motifs Membrane spanning (EMBOSS: tmap, topo) Signal peptide (EMBOSS: sigcleave) Coiled coil (EMBOSS: pepcoil) Helix-turn-helix (EMBOSS: helixturnhelix) Predicted from abundance of specific amino acids in a window and patterns of hydrophobic/hydrophillic “Super-secondary” Structure
Predict Protein server: : EMBL Heidelberg http://www.embl-heidelberg.de/predictprotein/ SOSUI: Tokyo Univ. of Ag. & Tech., Japan http://www.tuat.ac.jp/~mitaku/adv_sosui/submit.html TMpred (transmembrane prediction): ISREC (Swiss Institute for Experimental Cancer Research) http://www.isrec.isb-sib.ch/software/TMPRED_form.html COILS (coiled coil prediction): ISREC http://www.isrec.isb-sib.ch/software/COILS_form.html SignalP (signal peptides): Tech. Univ. of Denmark http://www.cbs.dtu.dk/services/SignalP/ Web servers that predict these structures
Protein Domains/Motifs • Proteins are built out of functional units know as domains (or motifs) • These domains have conserved sequences • Often much more similar than their respective proteins • Exon splicing theory (W. Gilbert) • Exons correspond to folding domains which in turn serve as functional units • Unrelated proteins may share a single similar exon (i.e.. ATPase or DNA binding function)
Protein Motif Databases • Known protein motifs have been collected in databases • Best database is PROSITE • The Dictionary of Protein Sites and Patterns • maintained by Amos Bairoch, at the Univ. of Geneva, Switzerland • contains a comprehensive list of documented protein domains constructed by expert molecular biologists • Alignments and patterns built by hand!
PROSITE is based on Patterns Each domain is defined by a simple pattern • Patterns can have alternate amino acids in each position and defined spaces, but no gaps • Pattern searching is by exact matching, so any new variant will not be found (can allow mismatches, but this weakens the algorithm) ID CBD_FUNGAL; PATTERN. AC PS00562; DT DEC-1991 (CREATED); NOV-1997 (DATA UPDATE); JUL-1998 (UPDATE). DE Cellulose-binding domain, fungal type. PA C-G-G-x(4,7)-G-x(3)-C-x(5)-C-x(3,5)-[NHG]-x-[FYWM]-x(2)-Q-C
Tools for Pattern searching EMBOSS • fuzznuc: DNA pattern search • fuzzpro: protein pattern search • preg: regular expression search of a protein sequence
Tools for PROSITE searches Free Mac program: MacPattern • ftp://ftp.ebi.ac.uk/pub/software/mac/macpattern.hqx Free PC program (DOS): PATMAT • ftp://ncbi.nlm.nih.gov/repository/blocks/patmat.dos EMBOSS has the programs: patmatdb, patmatmotifs Also in virtually all commercial programs: MacVector, VectorNTI, CLC-Bio, LaserGene, etc.
Websites for PROSITE Searches ScanProsite at ExPASy: Univ. of Geneva • http://expasy.hcuge.ch/sprot/scnpsit1.html Network Protein Sequence Analysis: Institut de Biologie et Chimie des Protéines, Lyon, France • http://pbil.ibcp.fr/NPSA/npsa_prosite.html PPSRCH:EBI, Cambridge, UK • http://www2.ebi.ac.uk/ppsearch/
Pattern Search Methods Complexity Consensus Pattern PSSM HMM Scores for each type of match in each position, gapped alignment exact match regular expression(defined mismatches) Position-specific gap scores fuzzy match Challenges to define statistical significance, sensitivity, & specificty What are all the true postives, & false negatives in a genome-wide search?
Profiles • Profiles are tables of amino acid frequencies at each position in a motif • They are built from multiple alignments • PROSITE entries also contain profiles built from an alignment of proteins that match the pattern • Profile searching is more sensitive than pattern searching - uses an alignment algorithm, allows gaps
Profile Alignment Gribskov et al. 1987 • Position specific scores • Allows addition of extra sequence(s) to an alignment • Allows alignment of alignments • Gaps introduced as whole columns in the separate alignments • Optimal alignment in time O(a2l2) a = alphabet size, l = sequence length • Information about the degree of conservation of sequence positions is included (similar amino acids)
Good reasons to use profile alignments • Adding a new sequence to an existing multiple alignment that you want to keep fixed(align sequence to profile) • Searching a database for new members of your protein family (pfsearch) • Searching a database of profiles to find out which one your sequence belongs to (pfscan) • Combining two multiple sequence alignments(profile to profile)
EMBOSS ProfileSearch • EMBOSS has a set of profile analysis tools. • Start with a multiple alignment • prophecy: create a profile • profit:scans a database with your profile • prophetmakes pairwise alignments between a single sequence and a profile
Websites for Profile searching • PROSITE ProfileScan: ExPASy, Geneva • http://www.isrec.isb-sib.ch/software/PFSCAN_form.html • BLOCKS (builds profiles from PROSITE entries and adds all matching sequences in SwissProt): Fred Hutchinson Cancer Research Center, Seattle, Washington, USA • http://www.blocks.fhcrc.org/blocks_search.html • PRINTS(profiles built from automatic alignments of OWL non-redundant protein databases): http://www.biochem.ucl.ac.uk/cgi-bin/fingerPRINTScan/fps/PathForm.cgi
More Protein Motif Databases • PFAM(1344 protein familyHMM profiles built by hand):Washington Univ., St. Louis • http://pfam.wustl.edu/hmmsearch.shtml • ProDom (profiles built from PSI-BLAST automatic multiple alignments of the SwissProt database): INRA, Toulouse, France • http://www.toulouse.inra.fr/prodom/doc/blast_form.html [This is my favorite protein database - nicely colored results]





















