Download

1 / 74
780 likes | 1.23k Views
Autosomal monogenic inheritance. Dr. habil . Kohidai Laszlo Department of Genetics , Cell- and Immunobiology Semmelweis University Budapest /2014/. Autosomal - Dominant. Minimum one of the parents is affected Phenotype of homozygotes is more severe than heterozygotes

E N D
Autosomalmonogenicinheritance Dr. habil. KohidaiLaszlo Department of Genetics, Cell- and Immunobiology Semmelweis University Budapest /2014/
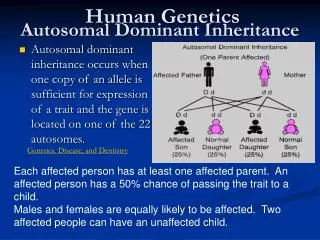
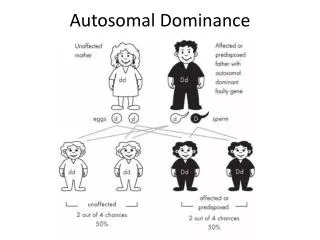
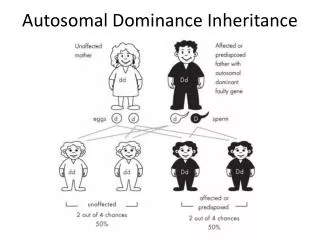
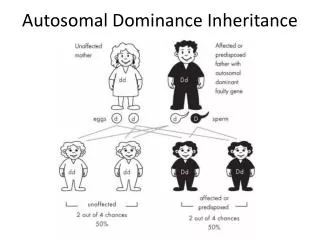
Autosomal - Dominant • Minimum one of the parents is affected • Phenotype of homozygotes is more severe than heterozygotes • Male and female are affected equally • Male and female transmit evenly • Affected x Non affected results 50%<affetced (sick)phenotype • Vertical pedigree • Frequency of the mutations shows correlation to the age of father • AD mutations influence receptor, structural or carrier proteins • Variable expressivity and penetrance
Dominant autosomal • ~ 2200 known dominant trait • frequency 0.1-3/1000/birth • most frequently affected organs: skeleton central nerve system
4p16.3 Achondroplasia Frequency 1:25000 • FGFR3 gene mutation • (fibroblast-growth • factor receptor 3) • Longitudinal growth of tubular bones is affected • Limbs are affected • forehead is dominant, middle part of the face is less developed
Rhinoceros unicornis sheeps Teleoceras fossiger
FGFR3 gene locus:4p16.3 • DNA: 16.5 Kb; 19 exon; exon 1 is not known in human • RNA: 4.0 Kb mRNS;alternativesplicing • exons 7 and 8: two mRNA isoforms IIIb and IIIc • Expressed in: brain, cartilage, liver, kidney, inner ear
The protein • 806 aa; 115 kDa • function: tyrosin kinase receptor • structure: extracellular part 3 Ig-like loops (I, II, III) strongly hydrophobe TM domain (22 aa) -TM intracellular domain with ttyrosine kinase activity -TK
Mutationsa of the FGFR3 gene 3 diseases are associated to the mutations of FGFR3
Arachnodactylia – Marfan syndrome Antoine Bernard-Jean Marfan (1896) Gabrielle
Arachnodactylia – Marfan syndrome Tutankhamen pharaoh Ehnaton pharaoh
Mary of Scotland Abraham Lincoln
Marfan syndrome – Symptomes • affected bones and joints • height • chest • long fingers • hyperflexibility
Eye and vision • myopia (short sight) • axis of the eye is longer • position of the lens is abnormál Heart and circulation • valve prolapse • aorta aneurysm • hypotension Marfan szindróma - Symptomes Frequency of mutations is increasing by age
15q21.1 There are several mutations of fibrillin gene (see green bands) Fibrillin protein • ~ 60 domain • binds 47 Ca2+ • similar to epidermal growth factor (EGF) Marfan syndrome Fibrillin gene (FBN1)
Osteogenesis imperfecta I. blue sclera Penetrance 100% extremely fragile bones Deafness or loss of hearing (penetrance is less than 100%) Level of pleiotropy is high
Osteogenesis imperfecta RNA: 2 RNA: 5.8 kb and 4.8 kb difference in 3’ UTR Protein :140 kDa 17q21.31-q22 COL1A1 gene • COL1A1 - 18 kb • 52 exon ( 6 – 49: alpha helical domain) • short exons: 45 bp, 54 bp or repeats of these two
Structure of collagen fibre Healthy Osteogenesis imp. Type I. Central helical domain: - 338 x repeat of Gly-X-Ytriplet - X and Y amino acids are frequently prolins (Pro)
Familiar hypercholesterinaemy • Main clinical symptoms: • early onset of cardial and • circulatory system diseases • (myocardial innfarction, • vascular diseases of brain and • peripherial blood vessels) • xanthoma • diseases of the eye
Familiar hypercholesterinaemy (FH) LDL lifespan in the body healthy: 2.5 days FH: 4.5 days LDL-level in sera is increased Reasons: - Mutation of LDL-receptor - ApoB defect LDL
Familiarhypercholesterinaemia Mutations of LDL-receptor Heterozygtes: 1:500-1000 Homozygotes: 1:1.000.000 Most frequent mutation: 9. exon 408 kodon CTG → CTA Val →Met
O-linked szénh.dom. Citopl. domain EGFP domain Ligand kötő domain Membrán Familiar hypercholesterinaemy Outcomes of LDL-receptor mutation Joseph Goldstein, Michael Brown (Nobel Prize 1985)
CAG trinucleotiderepeats Number of CAG repeats: Normal - >26 Transient 27-35 Low penetrance 36-39 High penetrance above 40 Huntington chorea • Starts in age 35-44 • Complex disease of locomotor, cognitive and psychiatric symptomes
4 Huntington chorea - (CAGn) • Gain-of-function mutation • The function of the Huntingtin gene in human is not known
Huntington choreaEffects of huntingtinongenelevel Inhibitedexpression of Dopamine D2 receptor gene
Huntington choreaEffectsoncytoskeletonlevel BDNF - brain-derived neurotrophic factor Transport of vesiclescontainingneurotransmitters viamicrotubularsystem: Huntingtin – huntingtin-related-protein (HAP) – dynactin - dynein
Correlation between the ‘CAG’ repeat-number and the age of onset
George Huntington (1850-1916) • Grandfather and father were farmer doctors – their anamnestic files supported Huntington to describe the disease • The disease was described in 1872 • Medical and Surgical Reporter of Philadelphia chorea = maniacdance
Anticipation – Trinucleotiderepeat The disease is expressed in gradually more severe levels and earlyier in the offspring generations
Sicklecellanemia Co-dominant • Haemoglobinopathy • HBB gene – 11 chrs. • b-globin chain mutation • HbS variant
Haemoglobin structural change HbA HbS
Duetotheirregularstructure of HbS: • The membraneof RBC is damaged • The affectedRBCsareeliminatedingreatnumbersintheperipheralorgans, (e.g. kidney/spleen) • The O2transport is disturbed
Malaria – spreading cycle Plasmodium falciparum Plasmodium vivax
Selective advantage of heterozygotes: Malaria Sicklecellanemia
Selective advantage of heterozygotes: Malaria Sickle cell anaemia BUT: The ratio is decreasing by changing the environment E.g. Cyprus – frequency of thalasszemia is decreasing
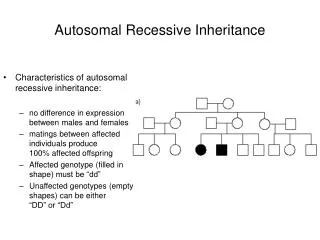
Autosomal Recessive • Parentsof theaffectedpersonusuallynotexpressingthetrait, theyareheterozygotes (Aa) • Expressionrateinmale:female is 1:1 • Transferredbymale and females • Co-saguinity of parentsis frequent • Risktohave an affectedindividualintheoffspring is 25% (Aa x Aa) • Horizontalpedigree
Recessive autosomal • 1700 known human recessive traits • More than 15% is enzymopathy (e.g. phenylalanine hydroxylase, hexose aminidase) • Mutations of haemoglobin Multiplex allelism – several mutations of one gene are responsible for the development of a symptome Complex heterozygote – an individual who possess two diverse, mutant allels of a gene mutáns allélját hordozza E.g. Cystic fibrosis – 850 different mutations
Frequency of recessive diseases There is a significant difference in diverse ethnic groups REASON:reproductive advantage of heterozygotes to homozygotes ENVIRONMENTAL FACTORS Selective advantages of heterozygotes Other reasons: directed marriages in some ethnic groups
b-Thalassemia (thalassa = sea)
b-Thalassemia (thalassa = sea) • haemoglobin b chain mutations • short life span of RBCs • O2 transporter capacity is decreased