Download

1 / 13
130 likes | 153 Views
Explore density-based classification of protein structures using iterative TM-score algorithm. Learn about feature vectors extraction, scoring, and experimental results. Enhance protein classification accuracy and speed.

E N D
Density-Based Classification of Protein Structures Using Iterative TM-score David Hoksza, Jakub Galgonek Charles University in PragueDepartment of Software Engineering Czech Republic
Presentation Outline • Biological background • Similarity search in protein structure databases • Method • feature vectors’ extraction • feature mapping • scoring • Experimental results • Conclusion CSBW 2009
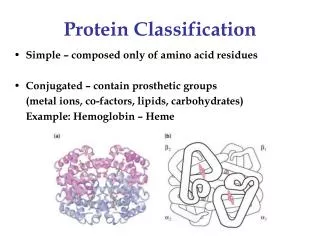
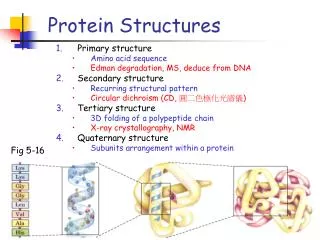
Biological Background • Proteins • molecule • translated from mRNA in ribosomes • DNA → RNA → protein • sequence of amino acids (20 AAs) • AAs coded by codons (triplet of nucleotides) • Function of a protein derived from its three dimensional structure • → similar proteins show similar functions • Identifying protein structure → finding similar proteins → getting clue to the function CSBW 2009
Similarity Search in Protein Databases • Similarity between a pair of proteins • finding mapping (alignment) of atoms • transformation to minimize mutual distance • computing distance of the superposed structures • DALI, CE, TM-align, Vorometric, Vorolign, PPM, … • Classification • SCOP (Structural Classification of Proteins) • manually curated hierarchical classification • family → superfamily → fold → class • need for automated classification CSBW 2009
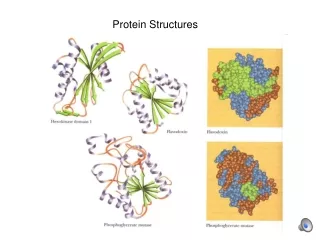
Feature Extraction and Comparison • Features based on density (inter-residual distances) of atoms • distances among Cα atoms used • each AA represents one feature→ protein p consists of |p| features • various semantics used • based on clustering Cα atoms into rings • distance between pair of features • Euclidean distance • weighted Euclidean distance CSBW 2009
Feature Extraction Semantics • Features • n-dimensional vectors of real numbers • AA ≈ viewpoint → VPT (viewpoint tag) • sDens • density of AAs in rings with a predefined width • sDensSSE • enhanced with SSE information • sRad • widths of rings containing predefined percentage of AAs • sRadSSE • enhanced with SSE information • sDir • number of AAs in a ring pointing from the viepoint • sDens enhanced with direction information CSBW 2009
Finding an Alignment • Modifications • Using modified distances of features in the substitution matrix • μ … mean of s[i,j] • Structure-specific gap costs • OGP … open gap penalty • EGP … extend gap penalty • Smith-Waterman dynamic programming solution (local alignment) • originally for sequence alignment • finding optimal local alignment according to given distance (substitution) matrix • L0,j = 0, Li,0 = 0 • L … DP matrix • s … distance matrix • σ … gap penalty • max(Li,j) … value of optimal alignment CSBW 2009
Superposition + Scoring • Scoring • Superposition achieved by iterative use of TM-score rotation algorithm • Algorithm tries to find a subset of alignment whose RMSD superposition maximizes the TM-score • Various initial subsets are evaluated • lenghts … L, L/2, max(L/25,4) • For each, a superposition is created and a new alignment consisting of spatially close residues is created • Iteration continues until stabilization or max. number of iteration is achieved • Superposition is used for reconstruction of the alignment achieved by dynamic programming (iteratively) CSBW 2009
Superposition Improvements Prefiltration using non-iterative FAST* heuristics top kNN filtering range filtering Belt-based restriction of dynamic programming when reconstructing original alignment Sticking more closely to the original alignment • Using only single initial alignment of length L • For similar proteins (which are important for classification) the deviation from original algorithm is small CSBW 2009
Experimental Results • ASTRAL compendium • Test set • ASTRAL 1.67 • only proteins not present in ASTRAL95 1.65 having < 30% sequence identity and <30 identical residues • 979 proteins • Database set • ASTRAL25 1.65 • 4357 proteins • Evaluation of • classification accuracy according to SCOP • classification of a query is based on the classification of the most similar protein • classification is correct when it is in agreement with SCOP classification • speed CSBW 2009
Comparison of Classification Methods CSBW 2009
Speed Evaluation CSBW 2009
Conclusion • We have proposed • Alignment of protein structures • distance and density of Cα atoms • alignment based on feature distances • Improved version of superposing algorithm (TM) • Accuracy • Speed • Experimental results • best results among methods according to SCOP superfamily and fold accuracy • 95.8% superfamily classification accuracy • 98.1% fold classification accuracy CSBW 2009