Download

1 / 35
360 likes | 370 Views
Array. An array can be considered as a multiply subscripted collection of data entries, for example numeric.

E N D
Array An array can be considered as a multiply subscripted collection of data entries, for example numeric. A dimension vector is a vector of non-negative integers. If its length is k then the array is k-dimensional. The dimensions are indexed from one up to the values given in the dimension vector. >array(c(1:100,500:400,700:800), c(10,10,3))
Matrix R allows simple facilities for creating and handling arrays, and in particular the special case of matrices. A matrix is a 2-dimensional array. >matrice <- array(1:12, dim=c(4,3)) >matrice >matrice <- matrix(1:12, ncol=3,nrow=4, byrow = TRUE) >matrice >matrice <- matrix(1:12, ncol=3,nrow=4, byrow = FALSE) >matrice >Colonna1<-1:4 >Colonna2<-5:8 >matrice<-cbind(Colonna1, Colonna2) >matrice >matrice<-rbind(Colonna1, Colonna2) >matrice
Lists An R listis an object consisting of an ordered collection of objects known as its components. There is no particular need for the components to be of the same mode or type, and, for example, a list could consist of a numeric vector, a logical value, a matrix, a complex vector, a character array, and so on. > lista<-list(DataFrame=fl2000,Carattere="Forte",Vettore=1:20) > lista
Data frames • A data frame is a list with class "data.frame". • There are restrictions on lists that may be made into data frames, namely: • The components must be vectors (numeric, character, or logical), factors, numeric matrices, lists, or other data frames. • Vector structures appearing as variables of the data frame must all have the same length, and matrix structures must all have the same row size. • A data frame may for many purposes be regarded as a matrix with columns possibly of differing modes and attributes.
Data Import/Export. Reading text data The easiest format has variable names in the first row case. In R, useremo Tabella <-read.table(”file.txt”,header=TRUE)
Data Import/Export. Sometimes columns are separated by commas (or tabs). ozone <- read.table("ozone.csv", header=TRUE,sep=",") Or ozone <- read.csv("ozone.csv")
Data Import/Export. Sometime the variable names aren’t included and you have to supply them. ozone <- read.table("ozone.csv", header=FALSE,sep=",”,col.names=c(“Ozone”,”Solar.R”,”Wind”,”Temp”,”Month”,”Day”))
Data Import/Export. Files for read.table can live on the web fl2000<-read.table("http://faculty.washington.edu/tlumley/data/FLvote.dat", header=TRUE)
Data Import/Export. The most common task is to write a matrix or data frame to file as a rectangular grid of numbers, possibly with row and column labels. Function write.table is more convenient, and writes out a data frame (or an object that can be coerced to a data frame) with row and column labels. >write.table(matrice,file=“Esercitazione_base.txt”, row.names = TRUE, col.names = TRUE)
Esercizi Create un vettore dimensione contenete i valori 4,4 ed un vettore numero contente i valori da 1 a 12. Creare una array mesi utilizzando il vettore dimensione e inserendo come dati i valori dell’oggetto month.name. Create una matrice mmesi 4 righe per 3 colonne inserendo come dati i valori dell’oggeto month.name per riga. Ripere il punto 3 inserendo i dati per colonna. Ripere il punto 3 inserendo come dati il vettore Misto<-c(month.name, numero ) Create un data.frame Dmesi usando gli oggetti month.name e numero. Create una lista contenete gli oggetti dimensione , mesi , mmesi. Importate I dati salvati nella cartella Esercizi in un nuovo data.frame.
Data Import/Export. Pdf(“file”) starts the graphics device driver for producing PDF graphics. It opens the file file and the PDF commands needed to plot any graphics requested are sent to that file. >Pdf(“esempioPDf.pdf”) >boxplot(fl2000) >dev.off()
Esercizi Create un file Mesi.txt nella cartella esercizi contenete i valori della matrice mmesi. Eliminare la matrice mmesi e ricrearla a partire dal file Mesi.txt. Creare un vettore Normale contenente 100 valori ottenuti da una distribuzione normale (rnorm) e produrre un file “normale.pdf” nella cartella Esercizi contenente i grafici plot(Normale ),hist(Normale), boxplot(Normale), e plot(density(Normale)) dev.off().
Bioconductor • Bioconductor is based primarily on the R programming language, but does contain contributions in other programming languages. • Bioconductor is an open source and open development software project to provide tools for the analysis and comprehension of genomic data. • The Bioconductor project was started in the Fall of 2001 and is overseen by the Bioconductor core team. It gained widespread exposure in the groundbreaking Genome Biology 2004 paper Bioconductor: open software development for computational biology and bioinformatics.
Bioconductor Packages • Most Bioconductor components are distributed as R packages, which are add-on modules for R. • Initially most of the Bioconductor software packages focused primarily on DNA microarray data analysis. As the project has matured, the functional scope of the software packages broadened to include the analysis of all types of genomic data, such as SAGE, sequence, or SNP data.
Install standard Bioconductor packages • Install BioConductor packages using the biocLite.R installation script. In an R command window, type the following: • >source("http://bioconductor.org/biocLite.R")>biocLite() • This installs the following packages: affy, affydata, affyPLM, annaffy, annotate, Biobase, Biostrings, DynDoc, gcrma, genefilter, geneplotter, hgu95av2.db, limma, marray, matchprobes, multtest, ROC, vsn, xtable, affyQCReport. After downloading and installing these packages, the script prints "Installation complete" and TRUE.
Load Packages • library and require load add-on packages. • library and require can only load an installed package. • >library(help = somename) computes basic information about the package. • >library() list all available packages. • Carichiamo la libreria affydata. • Carichiamo un dataset di esempio data(Dilution). Dilution
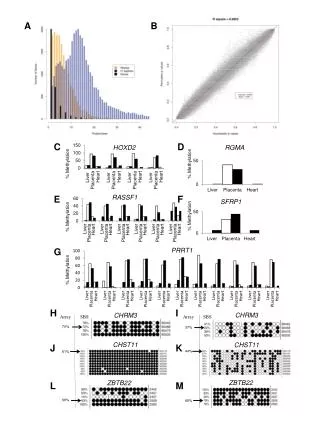
Section 1: Individual array quality The image plot • La funzione image() può essere usata per rilevare effetti spaziali dell’ibridazione. • >image(Dilution) creates an image for each sample.
Esercizi • Applicare la funzione pm() all’oggetto Dilution • Applicare le funzione mm(),probeNames(),sampleNames() all’oggetto Dilution. • Calcolare il numero delle probe di 20A per cui il mismatch è inferiore al perfect match. • Calcolare il valore medio delle probe di 20A per cui il mismatch è inferiore al perfect match usando la funzione mean. • Ripetere i punti 3 e 4 per le 10A. • Calcolare in numero delle probe di 20A per cui il mismatch è superiore al perfect match e che non sono presenti nel campione 10A con perfect match superiore al mismatch. • Creare un file Dilution.pdf nel quale dirigere l’output del comando images.
Section 2: Individual array quality The M vs A plot • The so-called M-A-plot is a graphical way to see ratios and fluorescence intensity at the same time. It was proposed by: Dudoit et al. Statistica Sinica (2002) 12:111. • As defined for two color cDNA arrays in that work: A = 1/2*(log2(Cy5) + log2(Cy3)) M = log2(Cy5 / Cy3) • For Affy arrays, • “Cy3” is the reference value, which is the median value of a pm over all chips chosen from a homogeneous group • “Cy5” is the pm value on the chip of interest • >MAplot(Dilution,pair=TRUE)
The M vs A plot >MAplot(Dilution,pair=TRUE) Typically, we expect the mass of the distribution in an MA plot to be concentrated along the M = 0 axis, and there should be no trend in the mean of M as a function of A
The M vs A plot >MAplot(Dilution,pair=TRUE) A trend in the lower range of A usually indicates that the arrays have different background intensities, this may be addressed by background correction. A trend in the upper range of A usually indicates saturation of the measurements, in mild cases, this may be addressed by non-linear normalisation (e.g. quantile normalisation).
The M vs A plot >MAplot(Dilution,pair=TRUE) A trend in the lower range of A usually indicates that the arrays have different background intensities, this may be addressed by background correction. A trend in the upper range of A usually indicates saturation of the measurements, in mild cases, this may be addressed by non-linear normalisation (e.g. quantile normalisation).
Boxplot • boxplots of the log2(Intensities). Each box corresponds to one array. It gives a simple summary of the distribution of probe intensities across all arrays. Typically, one expects the boxes to have similar size (IQR) and y position (median). • If the distribution of an individual array is very different from the others, this may indicate an experimental problem. After normalisation, the distributions should be similar. • >boxplot(Dilution)
Quality assessment with arrayQualityMetrics The function arrayQualityMetrics can be used on AffyBatch for Affymetrix data sets, ExpressionSet in the case of non Affymetrix one colour experiments and NChannelSet for dual colour experiments. arrayQualityMetrics produces a HTML report as an output. >library("arrayQualityMetrics") >arrayQualityMetrics(expressionset = CCl4, outdir = "CCl4", force = TRUE, do.logtransform = FALSE,split.plots = FALSE)
AffyRNAdeg • Uses ordered probes in probeset to detect possible RNA degradation. Plots and statistics used for evaluation. • Within each probeset, probes are numbered directionally from the 5' end to the 3' end. Probe intensities are averaged by probe number, across all genes. • >deg<-AffyRNAdeg(Dilution) • >plotAffyRNAdeg(deg)
Esercizi • Creare un file eserciziografici.pdf nel quale dirigere l’output dei seguenti esercizi. • Creare i seguenti grafici boxplot dei soli dati pm,dei soli dati mm e dei dati pm – mm. • Creare i grafici image dei valori di logartmo in base 2 dei perfect mach e dei mismatch. • Creare il grafico AffyRNAdeg per l’oggetto colorando di rosso i campioni 10A,20A e di verde i campioni 10B,20B. • Salvare in un file Eserciziopm.txt (cartella Esercizi) i valori di perfect match. • Applicare la funzione normalize() all’oggetto Dilution e salvarla nell’oggetto normalized_Dilution • Creare i grafici (se arrayQualityMetrics non funziona limitarsi a Boxplot e Maplot) dell’oggetto normalized_Dilution.
Esercizi • Scaricare da moodle il file Gene_list_ex1.txt ed importarlo in R con il comando read.table assegnandolo all’oggetto Lista_geni1. • Da un web browser connettersi al sito http://david.abcc.ncifcrf.gov/summary.jsp ed inserire il file Gene_list_ex1.txt come file Enter Gene List . Selezionare il miglior “Select Identifier” • Utilizzando il Functional Annotation Tool recuperare le informazioni reltive a GOTERM_BP_FAT, GOTERM_CC_FAT, GOTERM_MF_FAT ed importarle dentro R in tre oggetti separati. • Utilizzando il Functional Annotation Table importare il risultato in un oggetto.
source("http://bioconductor.org/biocLite.R") • biocLite("ShortRead") • The ShortRead package aims to provide key functionality for input, quality assurance, and basic manipulation of ‘short read’ DNA sequences such as those produced by Solexa, 454, and related technologies, including flexible import of common short read data formats. • exptPath <- system.file("extdata", package="ShortRead") • sp <- SolexaPath(exptPath) • class: SolexaPath experimentPath: /private/tmp/RtmpFPhpyj/Rinst67ac11ab7590/ShortRead/extdata • dataPath: Data • scanPath: NA • imageAnalysisPath: C1-36Firecrest • baseCallPath: Bustard • analysisPath: GERALD
readAligned: reading aligned data into R Solexa s_N_export.txt files (_N_ is a placeholder for the lane identifier) represent one place to start working the short read data in R. These files result from running ANALYSIS eland extended in the illumina sequencer. The files contain information on all reads, including alignment information for those reads successfully aligned to the genome. ShortRead parses additional align-ment files, including MAQ binary and text (mapview) files and Bowtie text files; • aln <- readAligned(sp, "s_2_export.txt") • aln • Class: AlignedRead • length: 1000 reads; • width: 35 cycles • chromosome: NM NM ... chr5.fa 29:255:255 • position: NA NA ... 71805980 NA • strand: NA NA ... + NA • alignQuality: NumericQuality • alignData varLabels: run lane ... filtering contig
Filtering input • Downstream analysis may often want to use a well-defined subset of reads. • These can be selected with the filter argument of readAligned. • There are built-in filters, for instance to remove all reads containing an N nucleotide, • to select just those reads that map to the genome file chr19.fa, • to select reads on the + strand, or to ‘level the playing field’ by selecting only a single read for any chromosome, position and strand: • nfilt <- nFilter() • cfilt <- chromosomeFilter('chr19.fa') • sfilt <- strandFilter("+") • ofilt <- occurrenceFilter(withSread=FALSE) • Here we select only those reads that map to chr19.fa: • chr19 <- readAligned(sp, "s_2_export.txt", filter=cfilt) • Filters can be ‘composed’ to act in unison, e.g., selecting only reads mapping to chr19.fa and on the + strand: • filt <- compose(cfilt, sfilt) • chr5plus <- readAligned(sp, "s_2_export.txt", filter=filt)
Filtering input Filters can subset aligned reads at other stages in the work flow, using a paradigm like the following: chr5 <- aln[cfilt(aln)] aln is an object of AlignedRead class. It contains short reads and their (calibrated) qualities: sread(aln) quality(aln)
Esercizi • Creare gli oggetti aln_min e aln_plus usando la funzione readAligned a partire dall’oggetto sp (sp <- SolexaPath(exptPath)) e la funzione strandFilter per lo strand – e +. • Usando la funzione alignData recuperare informazioni sulla • Lane usata nell’oggetto aln_min e aln_plus . • Usando la funzione position indicare quante reads hanno NA come posizione nell’oggetto aln_mins e nell’oggetto aln_plus. • Le reads che passano i criteri di filtraggio illumina contengono questa informazione nello slot data filtering (Y per reads che passano il filtro e N per quelle che non lo passano). Indicare in un oggetto count_plus_chr5 quate reads passano il filtro e sono relative la chr5. • Ripetere l’esercizio precedente per lo strand minus.
Quality assessment • The qa function provides a convenient way to summarize read and alignment quality. • One way of obtaining quality assessment results is • qaSummary <- qa(sp) • The qa object is a list-like structure. As invoked above and currently implemented, • qa visits all s_N_export.txt files in the appropriate directory. It extracts useful information from the files, and summarizes the results into a nested list-like structure. • Esercizio • Usare la funzione report per l’oggetto qaSummary .