Download

1 / 50
500 likes | 764 Views
Protein Folding/Unfolding. U (I ... I) N. The Folding Pathway(s). U, unfolded - I, intermediate - N, native. Assembly of proteins from building blocks. Packing of the secondary structure

E N D
Protein Folding/Unfolding
U (I ... I) N The Folding Pathway(s) U, unfolded - I, intermediate - N, native
Assembly of proteins from building blocks Packing of the secondary structure The major driving force for the folding of proteins appears to be the burying and clustering of hydrophobic side chains to minimize their contact with water: the “hydrophobic effect”. Structure and Mechanism in Protein Science A. Fersht (1999) WH Freeman and Company, New York, USA
Mechanisms for protein folding The diffusion-collision mechanism. Some micro-domains are formed prior collapse. Local elements of native secondary structure are formed independently of tertiary structure. The collapse-reorganization mechanism. NO microdomains formation prior collapse. Secondary and tertiary structures are formed in parallel. A. Fersht (1999): It is unlikely that there is a single mechanism for protein folding
Folding of an all-beta model protein The diffusion-collision mechanism The collapse-reorganization mechanism Y. Zhou, State University of New York at Buffalo, USA http://www.smbs.buffalo.edu/phys_bio
50 60 90 The diffusion-collision mechanism Some microdomains are formed prior collapse 1 20 33
55 70 90 The collapse-reorganization mechanism NO beta-sheets formation prior collapse 1 20 35
Time course of protein unfolding and refolding refolding unfolding Laser-heating T-jump of a three-helix bundle protein to 25 °C Experimental data after 800 ns are well fitted to a double exponential U. Mayor et al. (2003) Nature 421, 863-867
Molecular Dynamics (MD) Simulation Representative structures from the molecular dynamics simulations. Snapshots for the transition-state (TS) ensembles identified from the wild-type (WT) simulations at different temperatures U. Mayor et al. (2003) Nature 421, 863-867
Molecular Dynamics (MD) Simulation Structures from the protein 225 °C denaturation simulation shown in reverse, to illustrate a probable folding pathway of the protein to reach the native (N) state U. Mayor et al. (2003) Nature 421, 863-867
Molecular Dynamics (MD) Simulation The complete folding pathway of a protein from nanoseconds to microseconds U. Mayor et al. (2003) Nature 421, 863-867
U (I ... I) TS N The “molten globule” states U, Unfolded - I, Intermediate - TS, Transition State - N, Native The “molten globule” states are partly folded intermediate states of proteins that are characterized by having few tertiary interactions, some secondary structure, and a fluctuating hydrophobic core and by being separated from the native state by a high activation energy. Structure and Mechanism in Protein Science A. Fersht (1999) WH Freeman and Company, New York, USA
The structure of a molten globule. (A) A molten globule form of cytochrome b562 is more open and less highly ordered than the native protein, shown in (B). Note that the molten globule contains most of the secondary structure of the native form, although the ends of the alpha helices are frayed and one of these helices is only partly formed.
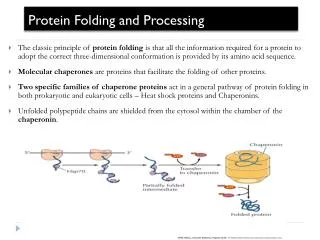
Molecular Chaperones in the Cytosol • To become functionally active, newly synthesized protein chains must fold to unique three-dimensional structures. • How this is accomplished remains a fundamental problem in biology. • Although the native fold of a protein is encoded in its amino acid sequence, protein folding inside cells is not generally a spontaneous process. • Many newly synthesized proteins require a complex cellular machinery of molecular chaperones and the input of metabolic energy to reach their native states efficiently. • The various chaperone factors protect nonnative protein chains from misfolding and aggregation, but do not contribute conformational information to the folding process. F.U. Hartl & M. Hayer-Hartl (2002) Science 295, 5561
Mechanisms of accelerated folding Confinement of non-native protein in the narrow, hydrophilic environment of the GroEL-GroES cage is suggested to result in a smoothing of the energy landscape (right), such that formation of certain trapped intermediates is avoided F.U. Hartl & M. Hayer-Hartl (2002) Science 295, 5561
Energy landscapes and modes of function of proteins (a) The `simplistic' model of proteins describes an energy landscape of a single stable conformer (i) and a function mode of either lock and key (ii) or induced fit (iii). (b) The `new view' assumes an ensemble of conformers of similar free energy (i), and a mode of function based on an equilibrium between two (or more) pre-existing isomers, only one of which exerts function (ii). LC James & DS Tawfik (2003) TIBS 28, 361-368
The co-evolution of fold and function (a) The enzyme is in equilibrium between different conformations. The native substrate (yellow) selects the dominant conformer (dark blue). (b) An alternative conformation potentiates the binding of a second substrate (pink). (c) Gene duplication enables one copy to evolve improved activity. LC James & DS Tawfik (2003) TIBS 28, 361-368
Protein folding. A newly synthesized protein rapidly attains a "molten globule" state. Subsequent folding occurs more slowly and by multiple pathways, some of which reach dead ends without the help of a molecular chaperone. Some molecules may still fail to fold correctly; these are recognized and degraded by proteolytic enzymes
Two families of molecular chaperones. The hsp70 proteins act early, recognizing small patches on a protein's surface. The hsp60-like proteins appear to act later and form a container into which proteins that have still failed to fold are transferred. In both cases repeated cycles of ATP hydrolysis by the hsp proteins contribute to a cycle of binding and release of the client protein that helps this protein to fold.
Molecular Chaperones in the Cytosol Models for the chaperone-assisted folding of newly synthesized polypeptides. TF, trigger factor; PFD, prefolding; NAC, nascent chain-associated complex F.U. Hartl & M. Hayer-Hartl (2002) Science 295, 5561
Molecular Chaperones in the Cytosol Chaperones that bind nascent chains: A) Structures of the ATPase domain and the peptide-binding domain of Hsp70 shown representatively for E. coli DnaK. B) Simplified reaction cycle of the DnaK system. C) Structure of archaeal PFD. F.U. Hartl & M. Hayer-Hartl (2002) Science 295, 5561
The GroEL-GroES chaperonin system A) Structure of the complex B) Simplified reaction of protein folding in the GroEL-GroES cage C) Mechanisms of accelerated folding Confinement of non-native protein in the narrow, hydrophilic environment of the GroEL-GroES cage is suggested to result in a smoothing of the energy landscape (right), such that formation of certain trapped intermediates is avoided F.U. Hartl & M. Hayer-Hartl (2002) Science 295, 5561
Molecular machines Schnitzer (2001) Nature 410, 878 - 881
Translocation machines - Mitochondria Once through the Toc complex, the pathway diverges. On the left is the pathway for proteins destined for the matrix. On the right is the pathway for import of polytopic inner membrane proteins. Chap: cytosolic chaperones 70: mitochondrial Hsp70 OM and IM: outer and inner membranes Outer translocon (Toc), in purple Inner translocon (Tic), in green Opening the door to mitochondrial protein import R.E. Jensen & A.E. Johnson (2001) Nature Struct Biol 8, 1008
Unfolding pathways of barnase a) during spontaneous unfolding in free-solution b) during import into mitochondria The parts of the structure shown in red unfold early, whereas those shown in blue unfold late. S. Huang et al. (1999) Nature Struct Biol 6, 1132
Unfoldingpathways of barnase Barnase is targeted tomitochondrial membranes by the addition of a mitochondrial presequence (solid black bar, lower picture). Unfolding and import requires an electrical membrane potential (DY) across the inner membrane and the ATP-dependent assistance ofmtHsp70 (hands) in the mitochondrial matrix D.N. Hebert (1999) Nature Struct Biol 6, 1084
Translocation machines - Mitochondria Outer-membrane proteins with a complicated topology pass through the TOM complex, then become integrated in the membrane with the assistance of a separate sorting and assembly complex (SAM). TOM, Outer translocon TIM, Inner translocon Tom20 is the major import receptor SAM, sorting and assembly complex Wiedemann et al. (2003) Nature 424, 565-571 Mihara (2003) Nature 424. 505-506
Translocation machines - Chloroplasts OM and IM: outer and inner membranes SPP: stromal processing peptidase C70, 60 and 93: chaperones Outer translocon (Toc), in green Inner translocon (Tic), in blue A GTPase gate for protein import into chloroplasts F. Kessler & D.J. Schnell (2002) Nature Struct Biol9, 81-83
Proteasome. Most of the proteins that are degraded in the cytosol are delivered to large protein complexes called proteasomes. Each proteasome consists of a central cylinder formed from multiple distinct proteases. Each end of the cylinder is "stoppered" by a large protein complex formed from at least 10 types of polypeptides, some of which hydrolyze ATP.
Toxic proteins in neurodegenerative diseases The ubiquitin-proteasome system. Proteins targeted for degradation are identified by covalent linkage to ubiquitin. Selective ubiquitination is accomplished by a series of enzymes (E1, E2, and E3) that constitute the ubiquitin ligase system. (B) Ubiquitinated substrates are recognized, unfolded, and degraded in an energy-dependent manner by the proteasome. JP Taylor et al. (2002) Science 296, 1991
Protein Aggregation • Large proteins often refold inefficiently, owing to the formation of partially folded intermediates that tend to aggregate. • Misfolding originates from interactions between regions of the folding polypeptide chain that are separate in the native protein. These nonnative states expose hydrophobic amino acid residues and readily self-associate into disordered complexes. • This aggregation process irreversibly removes proteins from their productive folding pathways, and must be prevented in vivo by molecular chaperones. • A certain level of protein aggregation does occur in cells and, in special cases, can lead to the formation of structured, fibrillar aggregates, known as amyloid, that are associated with diseases such as Alzheimer's or Huntington's disease F.U. Hartl & M. Hayer-Hartl (2002) Science 295, 5561
Molecular Chaperones in the Cytosol Aggregation of nonnative protein chains as a side-reaction of productive folding in the crowded environment of the cell. F.U. Hartl & M. Hayer-Hartl (2002) Science 295, 5561
Aggregation of misfolded proteins in neurodegenerative diseases (A) Alzheimer's disease. Arrow, extracellular amyloid plaque. (B) Fibrillar tau inclusions in Pick's disease. (C) PrPSc amyloid deposition in prion disease. (D) Multiple Lewy bodies in a nigral neuron in Parkinson's disease. (E, F) Neuronal intranuclear inclusions of mutant ataxin-3 in Machado-Joseph's disease. JP Taylor et al. (2002) Science 296, 1991
Medicine: Danger — misfolding proteins Protein folding is vital to living organisms. But errors in this process generate misfolded structures that can be lethal. R.J. Ellis & T.J.T. Pinheiro (2002) Nature 416, 483
APP A Secretasas b y g Agregación anormal g Placas seniles membrana La enfermedad de Alzhemier Teoría baptista: El corte aberrante de la proteína precursora del amiloide (APP) mediante dos proteasas (las secretasas beta y gamma) da lugar al fragmento beta-amiloide (Ab, cuya agregación origina la placa senil. La secretasa alfa corta APP de modo normal. J. Ávila, Diario de Sevilla, 11 Enero 2001
Alzheimer's and amyloid Amyloid-b peptides (Ab) come in a variety of sizes, of which the 42-amino-acid form (Ab42) is thought to contribute significantly to the development of Alzheimer's disease. B. Strooper&G. König (2002) Nature 414, 159
Quinasas Agregación Tau fosforilada Tau Ovillos neurofibrilares La enfermedad de Alzhemier Teoría taoísta: La proteína Tau ayuda a mantener el armazón estructural de las neuronas. Cuando se fosforila, la proteína Tau se agrega y aparecen los ovillos neurofibrilares, haciendo que las neuronas cambien de forma y dejen de funcionar. J. Ávila, Diario de Sevilla, 11 Enero 2001
Alzheimer's and amyloid Strategies for reducing the levels of amyloid peptides in the body. All have been validated in animal models. B. Strooper&G. König (2002) Nature 414, 159
Amyloid diseases Many human disorders — a well-known example being Alzheimer's disease — are characterized by the misfolding and aggregation of key proteins Structure of the pentamer of the serum amyloid P protein (SAP) L. Iversen (2002) Nature 417, 231
Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis This palindromiccompound (CPHPC) crosslinks and dimerizes SAP molecules. The normal plasma protein serum amyloid P component (SAP) binds to fibrils in alltypes of amyloid deposits, and contributes to the pathogenesis of amyloidosis. M.B. Pepyset al. (2002) Nature 417, 254
Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis Two SAP pentamers crosslinked byfive molecules of CPHPC M.B. Pepyset al. (2002) Nature 417, 254
Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis Whole-body 123I-labelled SAP scintigraphy. At 6 h, the blood pool background is completely absent and the liver, which is the only site of catabolism of SAP in vivo, has taken up the tracer. M.B. Pepyset al. (2002) Nature 417, 254
Possible routes of the BSE prion from cows to the human brain
Disulfide Bridge (Cys179 – Cys214) Prion replication and spread Normal Human Prion Protein (PrPC) (PDB entry 1QLX)
Prion replication and spread Cys179 Cys214 Oxidized Prion Protein (PDB entry 1QLX) Reduced Prion Protein (PDB entry1I4M)
Prion replication and spread Dimerization of Human Prion Protein The dimer results from the 3D swapping of the C-terminal helix 3 and rearrangement of the disulfide bond
Prion replication and spread Speculative model for conversion of PrPC to a PrPRDX fibril Lee & Eisenberg ( 2003) Nature Struct. Biol. 10, 725 - 730