Download

1 / 28
430 likes | 1.38k Views
Disorders of gonadal development. Gonadal sex Gonads or where gametes are produced by meiosis Somatic sex They can be divided into primary and secondary characteristics Secondary somatic sex characteristics are divided further into hair and body. FOR HUMAN MALES. Gonadal sex

E N D
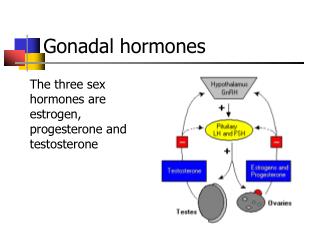
Gonadal sex Gonads or where gametes are produced by meiosis • Somatic sex They can be divided into primary and secondary characteristics Secondary somatic sex characteristics are divided further into hair and body
FOR HUMAN MALES • Gonadal sex Consists of the testes where the gametes or sperm are produced • Somatic sex characteristics Primary and Secondary • Male primary somatic sex characteristics • penis and scrotum • Male secondary somatic sex characteristics • Hair facial hair chest hair body hair • Body pelvic build straight hips muscular build upper body Ability to generate muscle mass at a faster rate than females following puberty
FOR HUMAN FEMALES • Gonadal sex Ovaries • Female primary somatic sex characteristics Clitoris, labia, vagina, cervix, uterus, fallopian tubes, and the ability to bear children • Female secondary somatic sex characteristics Hairvellus rather than terminal hair Body rounded hips, breasts, ability to nurse offspring, menstrual cycle, increased body fat composition, decreased upper body strength, decreased ability to generate muscle mass at a fast rate
Sexual development At the beginning of human development either male or female development is possible. • An embryo develops as a male or female using information from the Y chromosome. Unspecialized gonads and two sets of reproductive ducts exist until 6th week of development.
SEX REVERSAL • During male meiosis a synaptonemal complex forms and a chiasmaoccurs between X and Y chromosomes. • This results in regular exchange of material between the tips of XP and YP • Any genes in this region are inherited as thought they are autosomal and that is why it is called the pseudoautosomal region PAR But rarely happens mistake and the exchange occurs in region of critical genes which plays major role in gonadal dysgenesis. • This mutant hybrid chromosome is called an X (TDF) chromosome • When it fertilizes an X bearing egg it results in a 46 XX (TDF) male • When the corresponding mutant y chromosome fertilizes an egg it results in a 46 XY female
SEX REVERSAL 1 • Karyotype is XX but this individual will develop as an XY male because of the presence of the testes determining factor gene (TDF) • Remember!!! Presence of testes, determines gonadal sex in males • Even though these individual will look male they will suffer from testicular atrophy or small testes and sterility
SEX REVERSAL 1 • In humans if there are 2 X chromosomes in a male germ line , it acts as a poison to the germ cells and kills them during meiosis • The gonadal sex of this individual will be male because they will have testes • But they can not make viable sperm
SEX REVERSAL 2 • When a sperm carrying a Y (delTDF) chromosome fertilizes an X bearing egg the result is an • XY(delTDF) zygote • This individual develops as female even though the karyotype is XY • Ovaries are reduced and eggs that are produced will not survive • Have a female build but little pubic or underarm hair
AndrogenInsensitivitySyndrome • A personwho is geneticallymale (has one X andone Y chromosome) is resistanttomalehormones (androgens). • !! Genotype; 46,XY (male) Phenotype: Female (really??) • As a result, theperson has someorall of thephysicalcharacteristics of a woman, despitehavingthegeneticmakeup of a man.
Causes • Androgen insensitivity syndrome (AIS) is caused by various genetic defects on the X chromosome that make the body unable to respond to the hormones responsible for the male appearance. The syndrome is divided into two main categories: • Complete AIS • Incomplete AIS
Symptoms • The complete form of the syndrome occurs in as many as 1 in 20,000 live births. • Complete androgen insensitivity prevents the development of the penis and other male body parts. The child born appears to be a girl. There may be: • A vagina but no cervix or uterus • At puberty, female secondary sex characteristics (such as breasts, very little armpit and pubic hair) develop, but menstruation and fertility do not. • Inguinal hernia with a testis that can be felt during a physical exam • Testes in the abdomen or other unusual places in the body
Persons with incomplete AIS may have both male and female physical characteristics. Many have partial closing of the outer vaginal lips, an enlarged clitoris, and a short vagina. • The degree of sexual ambiguity varies widely in persons with incomplete AIS.; • breast development in men, • failure of one or both testes fail to descend into the scrotum after birth, and • hypospadias, a condition where the opening of the urethra is on the underside, rather than at the tip, of the penis. • Also included in the broad category of incomplete AIS is infertile male syndrome, which is sometimes due to an androgen receptor disorder.
Diagnosis Genetic: Chromosome analysis FISH studies Molecular analysis
Congenital Adrenal Hyperplasia • Congenital adrenal hyperplasia is a group of autosomal recessive disorders resulting from the deficiency of the enzymes required for the synthesis of cortisol in the adrenal cortex. 5 major enzymes: • 21-hydroxylase (%85) • 11-β-hydroxylase (%10) • 17-α-hydroxylase • 3-β-steroid hydrogenase • 20.22 desmolase
The most frequent is steroid21-hydroxylasedeficiency, accounting for more than 80 percent of cases. Steroid 21-hydroxylase (CYP21, also termed CYP21A2 and P450c21) is a cytochrome P-450 enzyme located in the endoplasmic reticulum. It catalyzes the conversion of 17-hydroxyprogesterone to 11-deoxycortisol, a precursor of cortisol, and the conversion of progesterone to deoxycorticosterone, a precursor of aldosterone.
Owing to this loss of enzyme function, patients with 21-hydroxylase deficiency cannot synthesize cortisol efficiently, and as a result, the adrenal cortex is stimulated by corticotropin and this causes excessive production of androgens.
Causes • Mutations in the CYP21(CYP21A2) gene. • CYP21 mutations can be grouped into three categories according to the level of enzymatic activity.
The first group consists of mutations such as deletions or nonsense mutations that totally ablate enzyme activity; these are most often associated with salt-wasting disease. (Classical form) • The second group of mutations, consisting mainly of the missense mutation, yields enzymes with 1 to 2 percent of normal activity • The final group includes mutations such as Val281Leu (V281L) and Pro30Leu (P30L) that produce enzymes retaining 20 to 60 percent of normal activity; these mutations are associated with the nonclassic disorder.
The classic form of CAH • The disorder usually manifests in childhood • Hypersecretion of adrenal androgens causes masculinization of the external genitalia of the female fetus • Affected infants can have ambiguous genitalia or even erroneous gender assignment • Because testicles are not present to produce müllerian inhibiting factor, the internal female organs are intact
Nonclassic (Mild) form of CAH Children Adults - Women • Syncope or near-syncope • Shortened stature compared with either parent • Hypotension (21-hydroxylase deficiency) • Hypertension (11-ß hydroxylase deficiency) • Clitorimegaly (mild) • Poorly developed labia • Hirsutism • Infertility • Polycystic ovary syndrome • Moderate to severe recurrent sinus or pulmonary infections • Severe acne • Hyperpigmentation, especially of the genitalia • Tall for age • Early onset of puberty
Diagnosis • Clinical ; Ambigious genitalia • Endocrinology: hormon profile enzyme • Genetical; Mutation analysis • Prenatal Diagnosis is possible, if there is an affected sib in the family, with a known mutation.
Treatment • There isn’t any gene therapy for the disease. • Endocrinologists have to follow-up for hormon therapy
There is a prenatal treatment of classic 21-OH forms of CAH. Dexamethasone (DEX) administration to the pregnant woman is used for the prevention of genital masculinization in female fetuses with CAH. DEX is a glucocorticoid which has a great suppressive effect on ACTH. So it inhibits the adrenal cortex through feedback on the hypothalamus and pituitary