Download

1 / 1
10 likes | 176 Views
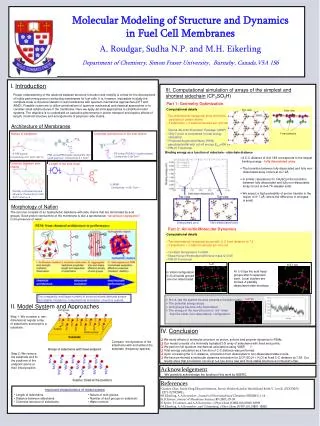
Ab initio Calculations of Interfacial Structure and Dynamics in Fuel Cell Membranes Ata Roudgar, Sudha P. Narasimachary and Michael Eikerling Department of Chemistry Simon Fraser University, Burnaby, BC Canada. 1. Introduction. 4. Proton Transform Mechanism at Interface.

E N D
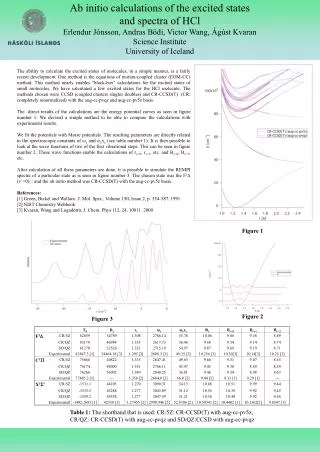
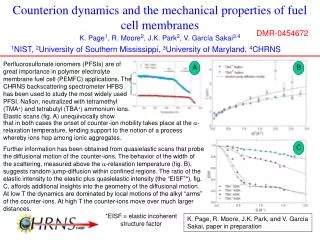
Ab initio Calculations of Interfacial Structure and Dynamics in Fuel Cell Membranes Ata Roudgar, Sudha P. Narasimachary and Michael Eikerling Department of Chemistry Simon Fraser University, Burnaby, BC Canada 1. Introduction 4. Proton Transform Mechanism at Interface Understanding the effect of chemical architecture, phase separation, and random morphology on transport properties and stability of polymer electrolyte membranes (PEM) is vital for the design of advanced proton conductors for polymer electrolyte fuel cells. Car-Parrinello Molecular Dynamics (CPMD) using functional BLYP Collective Coordinates and Minimum Reaction Path • Low temperature (T<100˚C), high degree of hydration, proton transfer in bulk, high conductivity Three collective coordinates: hydronium motion r, surface group rotation j and surface group tilting q. • High temperature (T>100˚C), low degree of hydration, proton transfer at interface, conductivity? DE= 0.55eV Evolution of PEM Morphology and Properties r q j Side view Top view Regular 10x10x10 grid of points is generated. Each point represents one configuration of the these three CCs. At each of these positions a geometry optimization including all remaining degrees of freedom is performed. The path which contains the minimum configuration energy is identified (as shown). 2 1 3 Frequency spectrum using AIMD Simulation • Car-Parrinello NVT simulation at T = 300K for upright conformation • Simulation time = 60ps • The frequency spectrum is calculated as a Fourier transform of velocity correlation function: j 2. Model of Hydrated Interfaces inside PEMs • Primary chemical structure • backbones • side chains • acid groups • Secondary structure • aggregates • array of side chains • water structure • Heterogeneous PEM • random phase separation • connectivity • swelling Focus on Interfacial Mechanisms of PT q hydrophobic phase Insight in view of fundamental understanding and design: • The fluctuations of sidechain rotation and sidechain tilting are responsible for proton transfer. • Low frequencies ≈ 100cm-1 are responsible for proton transfer. Feasible model of hydrated interfacial layer Self-organization into aggregates and dissociation 5. Proton Transform from Interface to Bulk hydrophilic phase Molecular interactions (polymer/ion/solvent), persistence length “Rescaled” interactions (fluctuating sidechains, mobile protons, water) Effective properties (proton conductivity, water transport, stability) Initialization of the second hydration shell • With this density we could make the second hydration shell consist of 14 water molecules. The surface group separation correspond to optimum density of water layer is dCC=7.07Å • The optimum density for one layer of water is calculated by varying the density of water layer. The average hydrogen bond length, <dO…O> = 2.92 Å Objectives • Correlations and mechanisms of proton transport in interfacial layer • Is good proton conductivity possible with minimal hydration? Assumptions: • decoupling of aggregate and side chain dynamics • map random array of surface groups onto 2D array • terminating C-atoms fixed at lattice positions • remove supporting aggregate from simulation Upright conformation Optimize geometry of minimally hydration and second hydration shell • The hydrogen bonds form in between water layer and oxygen atoms of Triflic acid • We calculated the binding energy between first and second hydration shells: • Ebin = ESG+wl – ESG – Ewl • The binding energy between first and second hydration shells as a function of dCC shows that for small dCC the second shell do not interact with minimally hydration Hydrophobic? • For large dCC the interaction between first and second shell binding energy is increased proton transform is more probable 3. Stable Structural Conformation Formation energy as a function of sidechain separation for regular array of Triflic acid, CF3-SO3-H Computational details highly correlated independent • Ab initio calculations based on DFT (VASP) • formation energy as a function of dCC • effect of side chain modification • binding energy of extra water molecule • energy for creating water defect 6. Conclusions • Correlations in interfacial layer are strong function of sidechain density. • Transition between upright (“stiff”) and tilted (“flexible”) configurations at dCC = 6.5Å involves hydronium motion, sidechain rotation, and sidechain tilting. • Reducing interfacial dynamics to the evolution of 3 collective coordinates enabled determination of transition path (activation energy 0.55 eV). • The binding energy of second shell becomes weak at small dcc No proton transfer from interface to bulk is expected. 2D hexagonal array of surface groups Unit cell: Side view Upon increasing sidechain there is a transition from “upright” to “tilted” structure occurs at dCC = 6.5Å dCC Upright Tilted fixed carbon positions • The tilted structure can be found in 3 different states: - fully dissociated - partially dissociated - non-dissociated • The largest formation energy E = -2.78 eV at dCC = 6.2 Åcorresponds to the upright structure. References • A. Roudgar, S. Narasimachary and M. Eikerling, J. Phys. Chem.B110, 20469 (2006). • A. Roudgar, S.P. Narasimachary, M. Eikerling .Chem. Phys. Lett. 457, 337 (2008) • M. Eikerling and A.A. Kornyshev, J. Electroanal. Chem. 502, 1-14 (2001). K.D. Kreuer, J. Membrane Sci. 185, 29- 39 (2001). • C. Chuy, J. Ding, E. Swanson, S. Holdcroft, J. Horsfall, and K.V. Lovell, J. Electrochem. Soc. 150, E271-E279 (2003). • E. Spohr, P. Commer, and A.A. Kornyshev, J. Phys.Chem. B 106, 10560-10569 (2002). • M. Eikerling, A.A. Kornyshev, and U. Stimming, J. Phys.Chem.B 101, 10807-10820 (1997).