Download
1 / 1
10 likes | 137 Views
Research Experience in Molecular Biotechnology & Genomics Summer 2007. Center for Integrated Animal Genomics. Characterization of novel WW domain containing genes that are expressed in L1 of the shoot apical meristem in maize. Jacqueline Heiss, Kazuhiro Ohtsu, and Patrick S. Schnable.

E N D
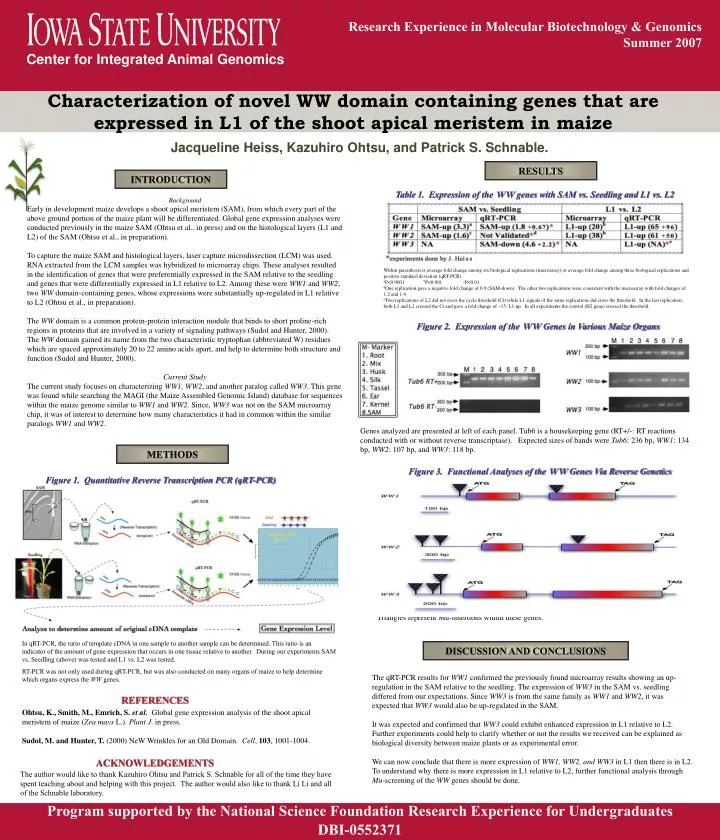
Research Experience in Molecular Biotechnology & Genomics • Summer 2007 Center for Integrated Animal Genomics Characterization of novel WW domain containing genes that are expressed in L1 of the shoot apical meristem in maize Jacqueline Heiss, Kazuhiro Ohtsu, and Patrick S. Schnable. RESULTS INTRODUCTION Table 1. Expression of the WW genes with SAM vs. Seedling and L1 vs. L2 Background Early in development maize develops a shoot apical meristem (SAM), from which every part of the above ground portion of the maize plant will be differentiated. Global gene expression analyses were conducted previously in the maize SAM (Ohtsu et al., in press) and on the histological layers (L1 and L2) of the SAM (Ohtsu et al., in preparation). To capture the maize SAM and histological layers, laser capture microdissection (LCM) was used. RNA extracted from the LCM samples was hybridized to microarray chips. These analyses resulted in the identification of genes that were preferentially expressed in the SAM relative to the seedling and genes that were differentially expressed in L1 relative to L2. Among these were WW1 and WW2, two WW domain-containing genes, whose expressions were substantially up-regulated in L1 relative to L2 (Ohtsu et al., in preparation). The WW domain is a common protein-protein interaction module that binds to short proline-rich regions in proteins that are involved in a variety of signaling pathways (Sudol and Hunter, 2000). The WW domain gained its name from the two characteristic tryptophan (abbreviated W) residues which are spaced approximately 20 to 22 amino acids apart, and help to determine both structure and function (Sudol and Hunter, 2000). Current Study The current study focuses on characterizing WW1, WW2, and another paralog called WW3. This gene was found while searching the MAGI (the Maize Assembled Genomic Island) database for sequences within the maize genome similar to WW1 and WW2. Since, WW3 was not on the SAM microarray chip, it was of interest to determine how many characteristics it had in common within the similar paralogs WW1 and WW2. Within parenthesis is average fold change among six biological replications (microarray) or average fold change among three biological replications and positive standard deviation (qRT-PCR). aP<0.0001 bP<0.001 cP<0.01 dOne replication gave a negative fold change of 0.9 (SAM-down). The other two replications were consistent with the microarray with fold changes of 1.2 and 1.4. eTwo replications of L2 did not cross the cycle threshold (Ct) while L1 signals of the same replications did cross the threshold. In the last replication, both L1 and L2 crossed the Ct and gave a fold change of ~15: L1-up. In all experiments the control (H2 gene) crossed the threshold. Figure 2. Expression of the WW Genes in Various Maize Organs Genes analyzed are presented at left of each panel. Tub6 is a housekeeping gene (RT+/-: RT reactions conducted with or without reverse transcriptase). Expected sizes of bands were Tub6: 236 bp, WW1: 134 bp, WW2: 107 bp, and WW3: 118 bp. METHODS Figure 3. Functional Analyses of the WW Genes Via Reverse Genetics Figure 1. Quantitative Reverse Transcription PCR (qRT-PCR) Triangles represent Mu-insertions within these genes. In qRT-PCR, the ratio of template cDNA in one sample to another sample can be determined. This ratio is an indicator of the amount of gene expression that occurs in one tissue relative to another. During our experiments SAM vs. Seedling (above) was tested and L1 vs. L2 was tested. RT-PCR was not only used during qRT-PCR, but was also conducted on many organs of maize to help determine which organs express the WW genes. DISCUSSION AND CONCLUSIONS The qRT-PCR results for WW1 confirmed the previously found microarray results showing an up-regulation in the SAM relative to the seedling. The expression of WW3 in the SAM vs. seedling differed from our expectations. Since WW3 is from the same family as WW1 and WW2, it was expected that WW3 would also be up-regulated in the SAM. It was expected and confirmed that WW3 could exhibit enhanced expression in L1 relative to L2. Further experiments could help to clarify whether or not the results we received can be explained as biological diversity between maize plants or as experimental error. We can now conclude that there is more expression of WW1, WW2, and WW3 in L1 then there is in L2. To understand why there is more expression in L1 relative to L2, further functional analysis through Mu-screening of the WW genes should be done. REFERENCES • Ohtsu, K., Smith, M., Emrich, S. et al. Global gene expression analysis of the shoot apical meristem of maize (Zea mays L.). Plant J. in press. • Sudol, M. and Hunter, T. (2000) NeW Wrinkles for an Old Domain. Cell, 103, 1001-1004. ACKNOWLEDGEMENTS The author would like to thank Kazuhiro Ohtsu and Patrick S. Schnable for all of the time they have spent teaching about and helping with this project. The author would also like to thank Li Li and all of the Schnable laboratory. Program supported by the National Science Foundation Research Experience for Undergraduates DBI-0552371