Download
1 / 1
10 likes | 72 Views
Enhance peptide identification sensitivity through PepArML meta-search engine, combining 7 search engines, machine learning, & spectral matching. Publicly available for free at Georgetown University.

E N D
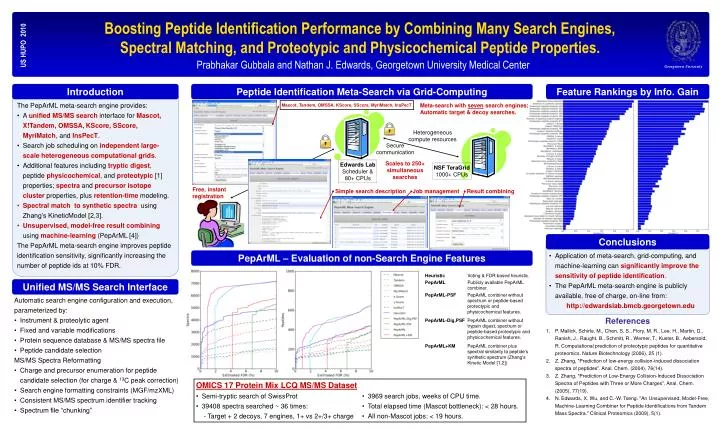
Georgetown University OMICS 17 Protein Mix LCQ MS/MS Dataset • Semi-tryptic search of SwissProt • 39408 spectra searched ~ 36 times: - Target + 2 decoys, 7 engines, 1+ vs 2+/3+ charge • 3969 search jobs, weeks of CPU time. • Total elapsed time (Mascot bottleneck): < 28 hours. • All non-Mascot jobs: < 19 hours. Boosting Peptide Identification Performance by Combining Many Search Engines, Spectral Matching, and Proteotypic and Physicochemical Peptide Properties. US HUPO 2010 Prabhakar Gubbala and Nathan J. Edwards, Georgetown University Medical Center Introduction Peptide Identification Meta-Search via Grid-Computing Feature Rankings by Info. Gain The PepArML meta-search engine provides: • A unified MS/MS search interface for Mascot, X!Tandem, OMSSA,KScore, SScore, MyriMatch, and InsPecT. • Search job scheduling on independentlarge-scale heterogeneous computational grids. • Additional features including tryptic digest, peptide physicochemical, and proteotypic [1] properties; spectra and precursor isotope cluster properties, plus retention-time modeling. • Spectral match to synthetic spectra using Zhang’s KineticModel [2,3]. • Unsupervised, model-free result combining using machine-learning (PepArML [4]) The PepArML meta-search engine improves peptide identification sensitivity, significantly increasing the number of peptide ids at 10% FDR. Meta-search with seven search engines;Automatic target & decoy searches. Mascot, Tandem, OMSSA, KScore, SScore, MyriMatch, InsPecT Heterogeneous compute resources Secure communication Scales to 250+ simultaneoussearches Edwards Lab Scheduler & 80+ CPUs NSF TeraGrid 1000+ CPUs Free, instantregistration Simple search description Job management Result combining Conclusions PepArML – Evaluation of non-Search Engine Features • Application of meta-search, grid-computing, and machine-learning can significantly improve the sensitivity of peptide identification. • The PepArML meta-search engine is publicly available, free of charge, on-line from: http://edwardslab.bmcb.georgetown.edu Unified MS/MS Search Interface • Automatic search engine configuration and execution, • parameterized by: • Instrument & proteolytic agent • Fixed and variable modifications • Protein sequence database & MS/MS spectra file • Peptide candidate selection • MS/MS Spectra Reformatting • Charge and precursor enumeration for peptide candidate selection (for charge & 13C peak correction) • Search engine formatting constraints (MGF/mzXML) • Consistent MS/MS spectrum identifier tracking • Spectrum file “chunking” References • P. Mallick, Schirle, M., Chen, S. S., Flory, M. R., Lee, H., Martin, D., Ranish, J., Raught, B., Schmitt, R., Werner, T., Kuster, B., Aebersold, R. Computational prediction of proteotypic peptides for quantitative proteomics. Nature Biotechnology (2006), 25 (1). • Z. Zhang, "Prediction of low-energy collision-induced dissociation spectra of peptides". Anal. Chem. (2004), 76(14). • Z. Zhang, "Prediction of Low-Energy Collision-Induced Dissociation Spectra of Peptides with Three or More Charges", Anal. Chem. (2005), 77(19). • N. Edwards, X. Wu, and C.-W. Tseng. "An Unsupervised, Model-Free, Machine-Learning Combiner for Peptide Identifications from Tandem Mass Spectra." Clinical Proteomics (2009), 5(1).






