Download

1 / 42
700 likes | 1.96k Views
Thrombosis, Embolism, Infarction. Bleeding and stopping bleeding. Extravasation of RBCs into tissues—not just fluids and solutes Enclosed bleeding: petechia, purpura, ecchymoses, hematoma

E N D
Bleeding and stopping bleeding • Extravasation of RBCs into tissues—not just fluids and solutes • Enclosed bleeding: petechia, purpura, ecchymoses, hematoma • Endothelin and tissue factor expressed by damaged vascular endothelium; circulating VII activated by tissue factor • Extrinsic, tissue activation of coagulation cascade • Circulating vWF and FXII exposed to ECM collagen • intrinsic, contact activation of coagulation cascade • Platelet adhesion and secretion • vasodilators, coagulation factors, growth factors, Ca++, ADP • Platelet aggregation and contraction • TxA2 synthesis by COX after Ca release in platelets • Coagulation cascade activates thrombin, which cleaves fibrinogen to fibrin • Fibrin polymerizes with factor XIII crosslinks to form a net that entraps RBCs and WBCs • Growth factors promote endothelial replacement • Plasmin digests fibrin after activation by tPA
Coagulation cascade highlights • Multiplicity of factors allows amplification or arrest • Factors named and numbered with roman numerals • Circulate as inactive, proenzyme or zymogen • Activated factors indicated with “a” • Most cascading factors synthesized in liver • Initiating factors synthesized by endothelium • Tissue factor (III), vWF • TxA2 synthesized in activated platelets; PGI2 synthesized in normal endothelium • Phospholipid surface of platelets and endothelium localizes cascade and anchors clot • Ca++ released from platelets crucial for platelet aggregation and initiation of coagulation cascade
Prostaglandins, COX, aspirin • Ca-activated PKC activates phospholipase A2, which liberates AA from cytoplasmic face of membrane • Arachidonic acid converted to prostaglandins by CycloOXygenase • Aspirin permanently inhibits COX I and II • Platelets cannot manufacture more COX, thromboxane synthesis stops • Endothelium expresses more COX to synthesize prostacyclin which promotes vasodilation, inhibits coagulation
Ca and vitamin K • Ca-dependent factors have g-carboxy glutamate residues in Ca-binding site • II (prothrombin), VII, IX, X • Post-translational modification catalyzed by g-glutamyl carboxylase with oxidation of vitamin K cofactor • Vitamin K epoxide reductase, (VKORC) reduces vitamin K back to its active form • Coumadin (warfarin) inhibits VKORC
Coagulation initiation in vivo • Extrinsic pathway almost immediately inhibited by epithelial TFPI, but thrombin is produced • Tissue Factor activates VII • VIIa converts X to Xa • Xa + Va prothrombinase produces thrombin • TFPI inhibits VIIa and Va • Thrombin activates common pathway • Thrombin activates VIIIa + IXa “tenase” complex that activates X • Thrombin activates XI, which in turn activates IX • Intrinsic pathway initiated by XII • Intrinsic pathway accelerated by thrombin positive feedback
Crucial complexes • Hageman factor, FXII, activated by interaction with negatively charged surfaces (collagen and HMWK) • FXIIa activates FXI and kallikrein • FXIa activates FIX, which forms “tenase” complex with FVIIIa to activate thrombin • Thrombin binds protease-activated receptors (e.g. PAR-1) on platelets, endothelial and smooth muscle cells, promoting inflammation
Activated Hageman Factor XIIa • Initiates inflammation and coagulation • Activates kallikrein, which produces vasoactive kinins • Activates the intrinsic clotting system, which induces formation of thrombin • Activates fibrinolysis by converting plasminogen to plasmin, creating inflammatory fibrinopeptides • Activates the complement system, specifically C3 and C5 anaphylatoxins
Deactivation of coagulation • Antithrombins • AT-III inhibits serine protease activity including thrombin, IXa, Xa, XIa, XIIa • AT-III activated by exogenous heparin and endothelial heparin-like molecules • Inhibition/inactivation of coagulation factors by proteins C and S • Plasminogen-plasmin fibrinolysis • tPA associated with fibrin plugs • urokinase-PA circulates in plasma
Antithrombotic properties of normal endothelium • Normal, intact vessels promote smooth, laminar blood flow • Formed components move in center of stream • Intact endothelial cells prevent exposure of ECM • Expression of ADPase (activates platelet GpIIa-IIIb interaction with fibrinogen) • Expression of thrombomodulin and heparin-like surface molecules • Expression of proteins C and S
Dysregulation of thrombosis • Epithelial damage • Initiates the clotting cascade • Turbulent or locally static blood flow: • Formed components contact vascular walls and ECM • Released clotting factors not transported or diluted • Inflammation activated • Hypercoaguability (thrombotic diatheses) • Inherited FV mutation doesn’t bind protein C
Causes of endothelial injury • Any perturbation in the dynamic balance prothombotic and antithrombotic activities of endothelium • Actual injury to vessels • Myocardial infarcts • Myocarditis • Cardiac jet lesions (abnormal flow) • Inflamed or prosthetic cardiac valves • Ruptured atherosclerotic plaques • Vasculitis syndromes • Radiation injury • High blood pressure • Cigarette smoke • Electrical injury • Invasion of vessel by tumor • Iatrogenic—physician caused
Altered blood flow • Turbulence • Ruptured atherosclerotic plaques • Arterial aneurysms • Dilated cardiac chambers (valve or muscle disease) • Vascular malformations • Stasis • Atrial fibrillation and other cardiac arrhythmias • Prolonged bed-rest or immobilization • Over-viscous blood (much more prone to stasis) • Sickle cell disease • Polycythemia (too much red cell mass) • Cryoglobulins (proteins that tend to precipitate)
Hypercoagulability • Inherited thrombophilia • Factor V Leiden • Q506R mutation prevents binding to protein C • Prothrombin SNIP causing increased transcription • (G20210A) in the 3′-untranslated region • Homocysteinemia • Homocysteine permanently degrades cysteine disulfide bridges and lysine amino acid residues in collagen, elastin, proteoglycans • Acquired thrombophilia • Nephrotic syndrome • Loss of small proteins including AT-III • Hyperestrogenemia • Increased hepatic coagulation factors • Aging • Decreased proeuction of endothelial PGI2
Heparin-induced thrombocytopenia • Administration of unfractionated heparin • Induces antibodies to heparin and heparin-like molecules on platelet and endothelial surfaces • Ab binding activates platelets • Platelets aggregate and are consumed • Antiphospholipid antibody syndrome • Autoantibodies to plasma proteins and coagulation factors • Recurrent thromboses • Repeated miscarriages • Cardiac valve vegetations • Thrombocytopenia
Grow retrograde to flow Begin at site of injury or turbulence Frequently occlusive Occur in coronary, cerebral, femoral arteries Grow with direction of flow Begin at site of stasis Always occlusive Occur in lower extremities 90%, also upper extremities, periprostatic plexus, ovarian or periuterine veins Arterial vs venous thrombi
Thrombi look like • Attached at one end and growing toward heart • Laminar in cross section when formed in flowing blood • Lines of Zahn represent layers of cell-free and cell-rich deposits • Red and solid when formed in static blood • Fatty and detached when formed post-mortem
Fate of thrombus • Propagation (continued formation) • Embolization • All or part dislodges and travels to other sites • Dissolution • Fibrinolysis mediated by consitutive tPA • Organization and recanalization • Ingrowth of endothelial and smooth muscle cells and fibroblasts with eventual re-establishment of capillary channels • Infection
Clinical consequences of thrombi • Superficial venous thrombi in saphenous veins • Varicose veins, edema, pain, infection • Deep vein thromboses • Associated with immobilization • Rapidly offset by collateral channels • Embolize to lungs • Arterial and cardiac thromboses • Predisposed by atherosclerosis • Embolize to brain, kidneys, spleen
Embolisms • Detached intravasular solid, liquid, gasous mass carried by the blood • Pulmonary embolisms • Often arise from deep vein thromboses • Associated with immobilization, hypercoagulability • Frequently small, silent, becoming organized • Right heart failure, cor pulmonale, when >60% pulmonary circulation obstructed • Rupture of obstructed arteries causes bleeding without infarction due to blood supply • Multiple emboli lead to hypertension and right ventricular failure
Systemic thromboembolism • Emboli in arterial circulation • Arise from intracardiac mural thrombi • 60% associated with left ventricular wall infarcts • 25% associated with atrial dilation or fibrillation • Remainder originate from aneurysms, valvular vegetation • Deposit in lower extremities or brain • Consequences depend on caliber of occluded vessel, redundant blood supply
Fat and marrow embolism • Release of fatty marrow from broken bones • Onset of symptoms 1 – 3 days after injury • Leads to pulmonary insufficiency • Tachypnea, dyspnea, tachycardia • Neurological symptoms • Irritability, restlessness • Thrombocytopenia • Platelets adhere to fat globules • Diffuse petechial rash
Fat and marrow embolism • Mechanical obstruction • Fat emboli with RBC and platelet aggregates occlude pulmonary and cerebral microvasculature • Biochemical injury • FFA released from fat globules injure endothelium initiating inflammation • Platelet aggregation and granulocyte recruitment result in free radicals, proteases, eicosanoids
Air embolisms • Iatrogenic consequences of • Coronary bypass surgery • Neurosurgery • Laparoscopic procedures • Chest wall injury • Decompression sickness • Nitrogen bubbles from blood within muscle, lungs, joints • Edema or ischemic necrosis in lungs (emphysema), femoral head, tibia, humerus
Amniotic fluid embolism • Infusion of amniotic fluid containing fetal components into uterine veins via rupture • Incidence 1:40K; mortality 80%; morbidity 13% total incidence, 85% survivors • Pulmonary microcirculation may contain • Fetal cells, vernix caseosa fat, fetal respiratory or GI mucin, lanugo hair • Onset characterized by sudden, severe dyspnea, cyanosis, shock, headache, seizures • Followed by pulmonary edema • Diffuse Intravascular Coagulation (DIC)
Infarction • Area of ischemic necrosis • Occlusion of either arterial supply or venous drainage • Thrombotic or embolic arterial occlusion • Venous occlusions tend to cause congestion and get bypassed • Myocardial infarction = heart attack • Cerebral infarction = stroke • Majority of US deaths
Infarcts look like • Red hemorrhagic infarcts • Venous occlusions • Congestion in loose tissues • In tissues with dual blood supply • lungs, small intestine, liver • Reperfusion of previously occluded flow • White anemic infarcts • Arterial occlusion • Solid organs • End-arterial circulation • heart, spleen, kidney
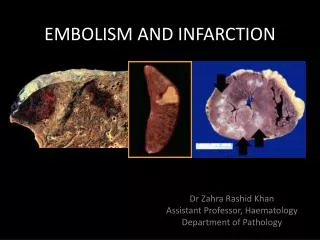
Coagulative necrosis—kidney infarction This is the typical pattern with ischemia and infarction (loss of blood supply and resultant tissue anoxia). Here, there is a wedge-shaped pale area of coagulative necrosis (infarction) in the renal cortex of the kidney. Microscopically, the renal cortex has undergone anoxic injury at the left so that the cells appear pale and ghost-like. There is a hemorrhagic zone in the middle where the cells are dying or have not quite died, and then normal renal parenchyma at the far right.
Infarctions in the spleen Infarction of many internal organs leads to a "pale" infarct with a wedge-shaped gross appearance (conical in 3 dimensions) from occlusion of a branching blood supply. Here are splenic infarcts in a patient with infective endocarditis. Portions of the vegetations have embolized to the spleen. These infarcts are typical of ischemic infarcts: they are based on the capsule, pale, and wedge-shaped. The remaining splenic parenchyma appears dark red.
Ischemic coagulative necrosis • Histological changes appear in 4 to 12 hours after anoxia • Cellular swelling, membrane degradation, nuclear condensation and breakdown • Inflammation is well defined in 1 to 2 days • Reparation follows • Labile tissues regenerate • Stable tissues scar
Determinants of infarct outcome • Nature of vascular supply • Dual blood supply or collateral vessels • Rate of development of occlusion • Slowly occluded vessels become organized with alternate perfusion pathways of collateral circulation • Vulnerability to hypoxia • Neurons, 3 – 4 minutes • Myocardial cells, 20 – 30 minutes • Fibroblasts, skeletal muscle, hours • Blood oxygenation • Anemia or cyanosis exacerbates hypoxia
Circulatory shock • Cardiogenic shock • low cardiac output due to myocardial pump failure • myocardial damage (infarction), ventricular arrhythmias, extrinsic compression (cardiac tamponade), or outflow obstruction (pulmonary embolism) • Hypovolemic shock • low cardiac output due to the loss of blood or plasma volume • massive hemorrhage or fluid loss from severe burns • May or may not be recoverable • Irreversible hypoxic injury to cells, tissues decrease odds of survival