Download

1 / 64
640 likes | 833 Views
Voltage-Gated Calcium Channels. “Excitable cells translate their electricity into action by Ca 2+ fluxes modulated by Voltage-Sensitive, Ca 2+ permeable channels.”. Brad Groveman Membrane Biophysics. Brief History.

E N D
Voltage-GatedCalcium Channels “Excitable cells translate their electricity into action by Ca2+ fluxes modulated by Voltage-Sensitive, Ca2+ permeable channels.” Brad Groveman Membrane Biophysics
Brief History • Discovered accidentally by Paul Fatt and Bernard Katz in neuromuscular transmissions in crab legs • Carbone and Lux termed LVA and HVA in in mammalian sensory neurons • Kurt Beam identified voltage-gated calcium channels as the voltage sensors in skeletal muscle
The Ion… Ca2+ Found in all Excitable Cells Shapes the Regenerative A.P. [Ca2+]i Three Best Studied Roles: 1. Contraction of Muscle 2. Secretion 3. Gating
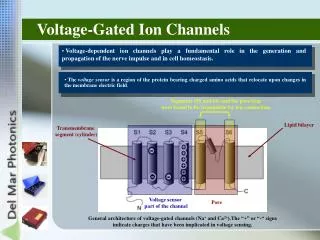
Structure/Function • Positively charged lysine and arginine residues in the S4 transmembrane segment thought to form the voltage sensor • Key negatively charged glutamate residues in each pore loop contributes to selectivity • Inactivation mechanism still unclear • [Ca2+]i elevation • Mode switching
Classes of VGCC http://calcium.ion.ucl.ac.uk/a1-nomenclature.html
Classes, Location, Blockers http://en.wikipedia.org/wiki/Voltage_gated_calcium_channel
Example Currents A. C. Dolphin 2006
Alpha-1 Subunit Structure http://calcium.ion.ucl.ac.uk/calcium-channels.html
Ribbon Structure of Alpha-1 http://calcium.ion.ucl.ac.uk/calcium-channels.html
Accessory Subunits http://calcium.ion.ucl.ac.uk/calcium-channels.html http://www.sigmaaldrich.com
Accessory Subunits • β - Contains Guanylate Kinase domain and SH3 domain • GK domain binds α1I-II intracellular loop • Stabilizes α1 and helps to traffic to membrane • Allows more current (higher amplitudes) for smaller depolarizations (HVA) • Shifts towards negative membrane potentials
Accessory Subunits • α2δ- co-expressed, linked by disulfide bond. • α2 extracellularly glycosylated • δ has a single transmembrane region • Co-expression enhances α1 expression • causes increased current amplitude, faster kinetics, and a hyperpolarizing shift in the voltage dependence of inactivation • Associates with all HVA calcium channels • Binding site for some anticonvulsant drugs
Accessory Subunits • γ- 4 transmembrane helices • Found in skeletal muscles • May have an inhibitory effect on calcium currents • Interact with AMPA and Glutamate receptors
Modulation • Upregulation of cardiac L-type channels by cyclic AMP-dependent protein kinase • Inhibitory modulation occurs via the activation of heterotrimeric G-proteins by G-protein-coupled receptors (GPCRs) • Calcium and Ca2+/CaM • Intracellular effector proteins (RyR, SNARE)
Synaptic Transmission • P/Q-types channels mainly responsible for transmitter release at central terminals • N-type channels prevalent in peripheral nerve terminals, responsible for synaptic transmission in autonomic and sensory terminals • L-type channels of the CaV1.3 and 1.4 class support synaptic transmission at specialized terminals • Continuous transmitter release in the retina and auditory hair cells with low depolarizations.
Pathologies • Neuropathic pain • Epilepsy • Congestive heart failure • Familial hemiplegic migraine • Several cerebellar ataxias
Important Domains • EF Hand Motif • Alloserically couples Ca2+ sensing apparatus with inactivation gate • Pre-IQ / IQ • Bind Calmudulin (Primary Ca2+ sensor) • Peptide A • Unknown Importance • ICDI • Inactivator of Calcium Dependent Inactivation • CaM1234 • CaM cant bind Ca2+
Inactivation • Typical fast channel inactivation conferred by voltage, but enhanced by Ca2+ feedback mechanism • Cav1.2 • Photoreceptors generate graded electrical response requires sustained Ca2+ influx • Seem to be devoid of CDI • Cav1.4 • major channel mediating Ca2+ influx in photoreceptors
Cav1.4 shows no CDI Ba2+ blocks CDI, focusing inactivation on voltage dependence f = Difference in normalized IBa and ICa remaining after 300ms of depolarization Cav1.2 shows typical “U” f curve Cav1.4 shows no difference Black – IBa Red – ICa
CaM binding in C-Terminal Proximal Distal CaM Binding In presence of Ca2+ No CaM Binding *Co-IP CaM1234 binding shows CaM binds Cav1.2 and 1.4 at basal Ca2+ conditions Loss of Calcium Sensor CaM NOT responsible for CDI insensitivity
C1884STOP *Modified CDI masked by inhibitory domain? Removal of last 100aa of Cav1.4 restored CDI but not Ba2+ inactivation Restored typical “U” shape voltage dependence and fmax nearly identical to Cav1.2
ICDI Domain C1884Stop co-expressed with CaM1234 Mutant to demonstrate that CDI is CaM dependent C1884Stop co-expressed with peptide of last 100aa to demonstrate presence of an inhibitory domain (ICDI) which is sufficient to block CDI effects * Red Box shows importance of sequence between aa1930 and aa1953 in CDI inhibition
Does ICDI interact with the Ca2+ sensing apparatus of Cav1.4? • Co-IP C-terminal fragments for interaction with ICDI • C-terminal fragments myc-tagged (IP) • IDCI Flag Tagged (IB) • ICDI IP with proximal C-terminal • IP abolished with deletion of EF hand motif • No interaction seen with peptides A or C from distal C-terminal
EF Hand target sequence for ICDI • GST-tagged IP of EF hand or EF hand with N-terminal Pre-IQ sequence • Both bound ICDI Target sequence • EF Hand motif and ICDI Domain both helical • Form paired helix which uncouples Ca2+ sensing apparatus from inactivation gate
Is inactivation of Cav1.2 rendered insensitive by Cav1.4 CT? Generated Cav1.2/1.4 Chimeras
Cav1.2/1.4 Chimeras demonstrate CDI inhibition • Inhibit CDI • Complete C-terminal replacement • C + ICDI replacement • A + ICDI replacement • Do No Block CDI • Addition of ICDI • Fusion of ICDI to IQ • Replacement of A • Peptide A and ICDI sufficient to abolish CDI • Peptide A does not bind ICDI Indirect
Proposed Model Gate opens Ca2+ interacts with CaM pre-bound to IQ motif causing conformational change in EF hand promoting interaction with channel conferring CDI ICDI constitutively binds EF hand impairing Ca2+/CaM induced conformation change. Inactivation strictly voltage-dependent with kinetics intrinsic to channel core
Pathophysiological Relevance • Loss of function mutation in Cav1.4 cause Congenital Stationary Night Blindness • Two mutations discovered in CSNB2 patients truncations in distal C-termial • Frameshift mutation identified in first 10aa of ICDI • All cause loss of ICDI function, allowing for CDI of photoreceptor Ca2+ channels
Chronic Hypoxia • Chronic Obstructive Pulmonary Disease • Arrhythmia • Stroke Reduction of Oxygen in brain
Previous Studies • APP expression increased following cerebral hypoxia or ischemia • Prolonged hypoxia enhances Ca2+ influx in PC12 cells apparently dependent on Aβ enhanced expression • Suggested Aβ composed Ca2+ pores as well as up-regulation of L-Type Ca2+ channels • THIS CANNOT BE EXTRAPOLATED TO CENTRAL NEURONS!!!
Mean Current Density vs Voltage RelationshipsCurrents based on VGCC • Current density in chronic hypoxic cells enhanced from normoxic conditions • Significantly at -10mV and 0mV • Inset shows no change in kinetics • Cd2+ non-selectively blocks VGCC • Abolished whole-cell Ca2+ current in both normoxic and hypoxic • Augmentation of current do to up-regulation of endogenous VGCC
Mean Current Density vs Voltage RelationshipsL-Type VGCC Responsible No difference seen in current under normoxic or hypoxic conditions in presence of L-Type Channel blocker Nimodipine Exaggerated difference seen in current under hypoxic conditions in presence of N-Type Channel blocker ω-CgTx
What does this have to do with APP? • Current augmentation caused by up-regulation in L-Type Ca2+ Channels • Immunohistochemical studies show increase in Aβ in hypoxic cells • This increase is abolished to normoxic conditions in presence of either γ or β-Secretase inhibitors
To beat a dead horse… • Hypoxia up-regulates L-Type Ca2+ Channels • Hypoxia increases Aβ production But are they related?
Blocking Aβ production by γ-Secretase inhibitorabolishes hypoxia effect Normoxic Hypoxic γ-Secretase inhibitor fully prevents Ca2+ currents augmentation by hypoxic conditions γ-Secretase inhibitor shows no effect on Ca2+ currents under normoxic conditions In presence of N-Type channel blockers
Blocking Aβ production by β-Secretase inhibitorabolishes hypoxia effect Normoxic Hypoxic β-Secretase inhibitor fully prevents Ca2+ currents augmentation by hypoxic conditions β-Secretase inhibitor shows no effect on Ca2+ currents under normoxic conditions In presence of N-Type channel blockers
Conclusions • Hypoxia increases formation of Aβ in primary culture neurons • Functional expression of L-Type Channels increased • Dependent on Aβ • Aβ do not form Ca2+ permeable pores
Status Epilepticus • Single episode can be evoked using chemical or electrical stimulation to mesial temporal lobe. <Pilocarpine> • Latent period of up to several weeks after first episode of normal behavior • Electrophysical changes including acquisition of low-threshold bursting behavior and high frequency clusters of 3-5 spikes
Bursting • Somatic bursting generated when spike after-depolarization (ADP) is large enough to attain spike threshold and trigger additional spikes • INaP currents drive bursting in ordinary cells • Intrinsic bursting in SE-experienced cells suppressed by Ni2+ Ca2+ driven • T-type Ca2+ channels (ICaT) implicated
Purpose • Contribution of ICaT vs ICaR • Subcellular localization of ICaT • Contribution of INaP
Bursting in early epileptogenesis driven by Ni2+ Sensitive Ca2+ Current “Jitters” seen in later spikes indicating a subthreshold ψR Small subthreshold hump Ni2+ suppresses bursting into single spike T-Type Ca2+ channels are blocked by Ni2+
ICaT vs ICaR • Ni2+ blocks both ICaT and ICaR • Previous studies show ICaT up-regulated after SE, but not ICaR • Cav3.2 T-type Ca2+ channel is 20-fold more sensitive to Ni2+ than other 2 splice variants • CaV3.2 provide critical depolarization for bursting
Amiloride suppresses bursting Blocks ICaT preferentially over HVA ICaR Also bock Na2+ exchangers Induces bursting by blocking KCNQ K+ Channels Bursting in normal cell not suppressed by Amiloride non-specific channel block not responsible for burst suppression
SNX-482 does not suppress bursting • Blocks ICaR • SNX-482 did not suppress bursting, however subsequent treatment with Ni2+ did • ICaR not critical, but is possibly auxiliary to bursting Ni2+ and Amiloride block bursting in SE cells, but SNX-482 does not ICaT Critical Bursting
INaP Contribution • PDB and Riluzole block INaPcompletely in pyramidal neurons without reducing transient Na+ currents • Subthreshold depolarizing potentials (SDP) also monitored • SDP blocked by TTX and INaP blockers, but not Ca2+ blockers INaP driven
SDP Reduced by PDB INaP blockage by PDB does not effect bursting, but reduces SDP to passive membrane response Subsequent addition of Ni2+ suppressed bursting