Download

1 / 110
1.11k likes | 1.32k Views
Lecture 15 February 8, 2010 Ionic bonding and oxide crystals. Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy. William A. Goddard, III, wag@wag.caltech.edu 316 Beckman Institute, x3093

E N D
Lecture 15 February 8, 2010 Ionic bonding and oxide crystals Nature of the Chemical Bond with applications to catalysis, materials science, nanotechnology, surface science, bioinorganic chemistry, and energy William A. Goddard, III, wag@wag.caltech.edu 316 Beckman Institute, x3093 Charles and Mary Ferkel Professor of Chemistry, Materials Science, and Applied Physics, California Institute of Technology Teaching Assistants: Wei-Guang Liu <wgliu@wag.caltech.edu> Ted Yu <tedhyu@wag.caltech.edu>
Course schedule Monday Feb. 8, 2pm L14 TODAY(caught up) Midterm was given out on Friday. Feb. 5, due on Wed. Feb. 10 It is five hour continuous take home with 0.5 hour break, open notes for any material distributed in the course or on the course web site but closed book otherwise No collaboration Friday Feb. 12, postpone lecture from 2pm to 3pm
Ring ozone Form 3 OO sigma bonds, but pp pairs overlap Analog: cis HOOH bond is 51.1-7.6=43.5 kcal/mol. Get total bond of 3*43.5=130.5 which is 11.5 more stable than O2. Correct for strain due to 60º bond angles = 26 kcal/mol from cyclopropane. Expect ring O3 to be unstable with respect to O2 + O by ~14 kcal/mol But if formed it might be rather stable with respect various chemical reactions. Ab Initio Theoretical Results on the Stability of Cyclic Ozone L. B. Harding and W. A. Goddard III J. Chem. Phys. 67, 2377 (1977) CN 5599
GVB orbitals of N2 VB view MO view Re=1.10A R=1.50A R=2.10A
The configuration for C2 Si2 has this configuration 2 1 0 1 2 1 2 4 3 4 4 2 2 From 1930-1962 the 3Pu was thought to be the ground state Now 1Sg+ is ground state 2 2
Ground state of C2 MO configuration Have two strong p bonds, but sigma system looks just like Be2 which leads to a bond of ~ 1 kcal/mol The lobe pair on each Be is activated to form the sigma bond. The net result is no net contribution to bond from sigma electrons. It is as if we started with HCCH and cut off the Hs
VB view MO view
The VB interference or resonance energy for H2+ The VB wavefunctions for H2+ Φg= (хL + хR) and Φu= (хL - хR) lead to eg = (hLL + 1/R) + t/(1+S) ≡ ecl + Egx eu = (hLL + 1/R) - t/(1-S) ≡ ecl + Eux where t = (hLR - ShLL) is the VB interference or resonance energy and ecl = (hLL + 1/R) is the classical energy As shown here the t dominates the bonding and antibonding of these states
Analysis of classical and interference energies egx = t/(1+S) while eux = -t/(1-S) Consider first very long R, where S~0 Then egx = t while eux = -t so that the bonding and antibonding effects are similar. Now consider a distance R=2.5 bohr = 1.32 A near equilibrium Here S= 0.4583 • = -0.0542 hartree leading to • egx = -0.0372 hartree while • eux = + 0.10470 hartree • ecl = 0.00472 hartree Where the 1-S term in the denominator makes the u state 3 times as antibonding as the g state is bonding.
Contragradience хL хR The above discussions show that the interference or exchange part of KE dominates the bonding, tKE=KELR –S KELL This decrease in the KE due to overlapping orbitals is dominated by tx = ½ [< (хL). ((хR)> - S[< (хL)2> Dot product is large and negative in the shaded region between atoms, where the L and R orbitals have opposite slope (congragradience)
The VB exchange energies for H2 For H2, the classical energy is slightly attractive, but again the difference between bonding (g) and anti bonding (u) is essentially all due to the exchange term. 1Eg = Ecl + Egx 3Eu = Ecl + Eux -Ex/(1 - S2) Each energy is referenced to the value at R=∞, which is -1 for Ecl, Eu, Eg 0 for Exu and Exg +Ex/(1 + S2)
Analysis of the VB exchange energy, Ex where Ex = {(hab + hba) S + Kab –EclS2} = T1 + T2 Here T1 = {(hab + hba) S –(haa + hbb)S2} = 2St Where t = (hab – Shaa) contains the 1e part T2 ={Kab –S2Jab} contains the 2e part Clearly the Ex is dominated by T1 and clearly T1 is dominated by the kinetic part, TKE. T2 T1 Ex Thus we can understand bonding by analyzing just the KE part if Ex TKE
Analysis of the exchange energies E(hartree) Eu1x Eg1x R(bohr) The one electron exchange for H2 leads to Eg1x ~ +2St /(1 + S2) Eu1x ~ -2St /(1 - S2) which can be compared to the H2+ case egx ~ +t/(1 + S) eux ~ -t/(1 - S) For R=1.6bohr (near Re), S=0.7 Eg1x ~ 0.94t vs. egx ~ 0.67t Eu1x ~ -2.75t vs. eux ~ -3.33t For R=4 bohr, S=0.1 Eg1x ~ 0.20t vs. egx ~ 0.91t Eu1x ~ -0.20t vs. eux ~ -1.11t Consider a very small R with S=1. Then Eg1x ~ 2t vs. egx ~ t/2 so that the 2e bond is twice as strong as the 1e bond but at long R, the 1e bond is stronger than the 2e bond
Noble gas dimers No bonding at the VB or MO level Only simultaneous electron correlation (London attraction) or van der Waals attraction, -C/R6 • LJ 12-6 Force Field • E=A/R12 –B/R6 • = De[r-12 – 2r-6] • = 4 De[t-12 – t-6] • = R/Re • = R/s where s = Re(1/2)1/6 =0.89 Re Ar2 s Re De
London Dispersion The weak binding in He2 and other noble gas dimers was explained in terms of QM by Fritz London in 1930 The idea is that even for a spherically symmetric atoms such as He the QM description will have instantaneous fluctuations in the electron positions that will lead to fluctuating dipole moments that average out to zero. The field due to a dipole falls off as 1/R3 , but since the average dipole is zero the first nonzero contribution is from 2nd order perturbation theory, which scales like -C/R6 (with higher order terms like 1/R8 and 1/R10)
London Dispersion The weak binding in He2 and other nobel gas dimers was explained in terms of QM by Fritz London in 1930 The idea is that even for a spherically symmetric atoms such as He the QM description will have instantaneous fluctuations in the electron positions that will lead to fluctuating dipole moments that average out to zero. The field due to a dipole falls off as 1/R3 , but since the average dipole is zero the first nonzero contribution is from 2nd order perturbation theory, which scales like -C/R6 (with higher order terms like 1/R8 and 1/R10) Consequently it is common to fit the interaction potentials to functional forms with a long range 1/R6 attraction to account for London dispersion (usually referred to as van der Waals attraction) plus a short range repulsive term to account for short Range Pauli Repulsion)
MO and VB view of He dimer, He2 MO view ΨMO(He2) = A[(sga)(sgb)(sua)(sub)]= (sg)2(su)2 VB view ΨVB(He2) = A[(La)(Lb)(Ra)(Rb)]= (L)2(R)2 Net BO=0 Pauli orthog of R to L repulsive Substitute sg = R +Land sg = R -L Get ΨMO(He2) ≡ ΨMO(He2)
Remove an electron from He2 to getHe2+ MO view Ψ(He2) = A[(sga)(sgb)(sua)(sub)]= (sg)2(su)2 Two bonding and two antibonding BO= 0 Ψ(He2+) = A[(sga)(sgb)(sua)]= (sg)2(su) BO = ½ Get 2Su+ symmetry. Bond energy and bond distance similar to H2+, also BO = ½
Remove an electron from He2 to getHe2+ - MO view Ψ(He2) = A[(sga)(sgb)(sua)(sub)]= (sg)2(su)2 Two bonding and two antibonding BO= 0 Ψ(He2+) = A[(sga)(sgb)(sua)]= (sg)2(su) BO = ½ Get 2Su+ symmetry. Bond energy and bond distance similar to H2+, also BO = ½ VB view Substitute sg = R +Land sg = L -R Get ΨVB(He2) ≡ A[(La)(Lb)(Ra)] - A[(La)(Rb)(Ra)] = (L)2(R) - (R)2(L)
He2+ 2Sg+ (sg)1(su)2 2Su+ (sg)2(su) BO=0.5 - + He2 Re=3.03A De=0.02 kcal/mol No bond H2 Re=0.74xA De=110.x kcal/mol BO = 1.0 MO good for discuss spectroscopy, VB good for discuss chemistry H2+ Re=1.06x A De=60.x kcal/mol BO = 0.5 Check H2 and H2+ numbers
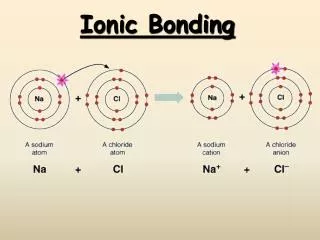
Ionic bonding (chapter 9) Consider the covalent bond of Na to Cl. There Is very little contragradience, leading to an extremely weak bond. Alternatively, consider transferring the charge from Na to Cl to form Na+ and Cl-
The ionic limit At R=∞ the cost of forming Na+ and Cl- is IP(Na) = 5.139 eV minus EA(Cl) = 3.615 eV = 1.524 eV But as R is decreased the electrostatic energy drops as DE(eV) = - 14.4/R(A) or DE (kcal/mol) = -332.06/R(A) Thus this ionic curve crosses the covalent curve at R=14.4/1.524=9.45 A Using the bond distance of NaCl=2.42A leads to a coulomb energy of 6.1eV leading to a bond of 6.1-1.5=4.6 eV The exper De = 4.23 eV Showing that ionic character dominates E(eV) R(A)
GVB orbitals of NaCl Dipole moment = 9.001 Debye Pure ionic 11.34 Debye Thus Dq=0.79 e
electronegativity To provide a measure to estimate polarity in bonds, Linus Pauling developed a scale of electronegativity () where the atom that gains charge is more electronegative and the one that loses is more electropositive He arbitrarily assigned =4 for F, 3.5 for O, 3.0 for N, 2.5 for C, 2.0 for B, 1.5 for Be, and 1.0 for Li and then used various experiments to estimate other cases . Current values are on the next slide Mulliken formulated an alternative scale such that M= (IP+EA)/5.2
Electronegativity Based on M++
Ionic crystals Starting with two NaCl monomer, it is downhill by 2.10 eV (at 0K) for form the dimer Because of repulsion between like charges the bond lengths, increase by 0.26A. A purely electrostatic calculation would have led to a bond energy of 1.68 eV Similarly, two dimers can combine to form a strongly bonded tetramer with a nearly cubic structure Continuing, combining 4x1018 such dimers leads to a grain of salt in which each Na has 6 Cl neighbors and each Cl has 6 Na neighbors
The NaCl or B1 crystal All alkali halides have this structure except CsCl, CsBr, CsI (they have the B2 structure)
The CsCl or B2 crystal There is not yet a good understanding of the fundamental reasons why particular compound prefer particular structures. But for ionic crystals the consideration of ionic radii has proved useful
Ionic radii, main group Fitted to various crystals. Assumes O2- is 1.40A NaCl R=1.02+1.81 = 2.84, exper is 2.84 From R. D. Shannon, Acta Cryst. A32, 751 (1976)
Role of ionic sizes in determining crystal structures Assume that the anions are large and packed so that they contact, so that 2RA < L, where L is the distance between anions Assume that the anion and cation are in contact. Calculate the smallest cation consistent with 2RA < L. RA+RC = (√3)L/2 > (√3) RA Thus RC/RA > 0.732 RA+RC = L/√2 > √2 RA Thus RC/RA > 0.414 Thus for 0.414 < (RC/RA ) < 0.732 we expect B1 For (RC/RA ) > 0.732 either is ok. For (RC/RA ) < 0.414 must be some other structure
Radius Ratios of Alkali Halides and Noble metal halices Rules work ok B1: 0.35 to 1.26 B2: 0.76 to 0.92 Based on R. W. G. Wyckoff, Crystal Structures, 2nd edition. Volume 1 (1963)
Radius rations B3, B4 The height of the tetrahedron is (2/3)√3 a where a is the side of the circumscribed cube The midpoint of the tetrahedron (also the midpoint of the cube) is (1/2)√3 a from the vertex. Hence (RC + RA)/L = (½) √3 a / √2 a = √(3/8) = 0.612 Thus 2RA < L = √(8/3) (RC + RA) = 1.633 (RC + RA) Thus 1.225 RA < (RC + RA) or RC/RA > 0.225 Thus B3,B4 should be the stable structures for 0.225 < (RC/RA) < 0. 414
Structures for II-VI compounds B3 for 0.20 < (RC/RA) < 0.55 B1 for 0.36 < (RC/RA) < 0.96
CaF2 or fluorite structure Like GaAs but now have F at all tetrahedral sites Or like CsCl but with half the Cs missing Find for RC/RA > 0.71
Rutile (TiO2) or Cassiterite (SnO2) structure Related to NaCl with half the cations missing Find for RC/RA < 0.67
CaF2 rutile CaF2 rutile
Electrostatic Balance Postulate For an ionic crystal the charges transferred from all cations must add up to the extra charges on all the anions. We can do this bond by bond, but in many systems the environments of the anions are all the same as are the environments of the cations. In this case the bond polarity (S) of each cation-anion pair is the same and we write S = zC/nC where zC is the net charge on the cation and nC is the coordination number Then zA = Si SI = Si zCi /ni Example1 : SiO2. in most phases each Si is in a tetrahedron of O2- leading to S=4/4=1. Thus each O2- must have just two Si neighbors
a-quartz structure of SiO2 Each Si bonds to 4 O, OSiO = 109.5° each O bonds to 2 Si Si-O-Si = 155.x ° Helical chains single crystals optically active; α-quartz converts to β-quartz at 573 °C rhombohedral (trigonal)hP9, P3121 No.152[10] From wikipedia
Example 2 of electrostatic balance: stishovite phase of SiO2 The stishovite phase of SiO2 has six coordinate Si, S=2/3. Thus each O must have 3 Si neighbors Rutile-like structure, with 6-coordinate Si; high pressure form densest of the SiO2 polymorphs tetragonaltP6, P42/mnm, No.136[17] From wikipedia
TiO2, example 3 electrostatic balance Example 3: the rutile, anatase, and brookite phases of TiO2 all have octahedral Ti. Thus S= 2/3 and each O must be coordinated to 3 Ti. top anatase phase TiO2 right front
Corundum (a-Al2O3). Example 4 electrostatic balance Each Al3+ is in a distorted octahedron, leading to S=1/2. Thus each O2- must be coordinated to 4 Al