Download

1 / 48
480 likes | 623 Views
Algorithmics of -1 frameshift RNA sequences. Michaël Bekaert 1 , Laure Bidou 1 , Alain Denise 1,2 , Guillemette Duchateau-Nguyen 1 , Céline Fabret 1 Jean-Paul Forest 2 , Christine Froidevaux 2 , Isabelle Hatin 1 , Jean-Pierre Rousset 1 , Michel Termier 1

E N D
Algorithmics of -1 frameshift RNA sequences Michaël Bekaert1, Laure Bidou1, Alain Denise1,2, Guillemette Duchateau-Nguyen1, Céline Fabret1 Jean-Paul Forest2, Christine Froidevaux2, Isabelle Hatin1, Jean-Pierre Rousset1, Michel Termier1 1 IGM (Institut de Génétique et Microbiologie) 2 LRI (Laboratoire de Recherche en Informatique) Université Paris-Sud, Orsay
Flow of genetic information DNAsequence replication CATATGGATTACATGGTCTAAGAT transcription RNAsequence CAU AUG GAU UAC AUG GUC UAA GAU translation Protein
Translation mRNA CAUAUGGAUUAC AUG GUCUAAGAU 5’ 3’
Translation ribosome CAUAUG GAUUAC AUG GUCUAAGAU 5’ 3’ The ribosome reads bases by triplets (or codons)from aSTART codon
Translation CAUAUGGAU UAC AUG GUCUAAGAU 5’ 3’ The ribosome synthetizes one amino-acid per codon
Translation CAUAUGGAU UAC AUG GUCUAAGAU 5’ 3’
Translation CAUAUGGAU UAC AUG GUCUAAGAU 5’ 3’
Translation CAUAUGGAU UAC AUG GUCUAAGAU 5’ 3’
Translation CAUAUGGAU UAC AUG GUCUAAGAU 5’ 3’
Translation CAUAUGGAU UAC AUG GUCUAAGAU 5’ 3’ The synthesis goes on until a STOPcodon is read 1 mRNA gives 1 protein
Experimental fact • Some mRNAs encode two distinct proteins with same beginning
STOP-1 START0 STOP0 0 phase ORF1a -1 phase ORF1b usual translation -1frameshift Programmed -1 frameshifting Non-deterministic event 1 mRNA gives 2 distinct proteinswith accurate ratio
Typical -1 frameshift site [Brierley, 1989] S2 3’ L1 L’1 S1 L2 5’ AUG NNXXXY YYZ P SP Secondary structure Slippery sequence
IBV frameshift site S2 U C C G A G C GAAA 3’ A G G C U C G G UGACGAUGGGG GCUG AUACCCC S1 5’ AUG UAU UUA AAC GGGUAC UUGC Pseudoknot Slippery sequence
Translation with frameshift U C C G A G C GAAA 3’ A G G C U C G G UGACGAUGGGG GCUG AUACCCC UUGC 5’ AUG UAUUUA AACGGG UAC
Translation with frameshift U C C G A G C GAAA 3’ A G G C U C G G UGACGAUGGGG GCUG AUACCCC UUGC 5’ UAU UUA AAC GGG UAC
-1 shift Translation with frameshift U C C G A G C GAAA 3’ A G G C U C G G UGACGAUGGGG GCUG AUACCCC UUGC 5’ UAU UUA AAC GGG UAC
Translation with frameshift 3’ 5’ UA UUU AAA CGG GUA CGG GGU AGC AGU
Translation with frameshift 3’ 5’ UA UUU AAA CGG GUA CGG GGU AGC AGU
Translation with frameshift 3’ 5’ UA UUU AAA CGG GUA CGG GGU AGC AGU
Translation with frameshift 3’ 5’ UA UUU AAA CGG GUA CGG GGU AGC AGU
Translation : mRNA & ribosome Adapted from Frank et al. by Giedroc et al.
Biological or randomsequences Model Wild-type foldedsequences Score matrix Folding Foldedsequences Mutantsequences Rules Voting Folded and sorted sequences New FS sites In silico andin vivovalidation
Search for FS sites: the easy part • Slippery sequence in -1 phase with START codon NNN N ATG NN XXX YYY Z
Search for FS sites :the not-so-easy part • Search of secondary structure Folding ? AGGACCT
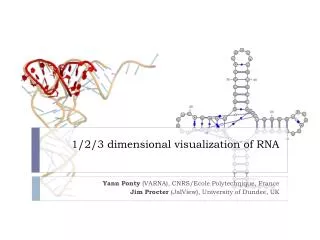
Example of a folded structure Picture from Lyngso and Pedersen 2000
Folding algorithms • Aligned sequences • Zuker’s • Rivas & Eddy’s
Algorithms that requirealigned sequences • Not relevant to our problem since we only fold one sequence at the same time
Folding using Zuker’s model • Tractable model based on additive energy minimization • One sequence gives one folding • Bases are either single-stranded or paired with a single other base • Matching interactions must not cross(i.e. pseudoknots are not allowed)
Base-pairs interactions nested disjoint crossing
Zuker’s algorithm • Does not find our pseudoknots, even if the two stems are looked for separately
Seeking pseudoknots • Rivas and Eddy 1999 • extends Zuker’s algorithm • accounts for pseudoknots using a more complex recursion (steep time and memory requirement) • does not work for our problem, probably due to lack of biological experiments to set the thermodynamical parameters
Orpheo • Seeks stems separately with adequate parameters
Score matrix A T C G A -6 2 -6 -6 T 2 -6 -6 0 C -6 -6 -6 4 G -6 0 4 -6
Smith-Waterman algorithm A G G A C C T A 0 0 0 0 0 2 G 0 0 4 6 0 G 0 10 4 0 A 0 0 2 C 0 0 C 0 T
Smith-Waterman algorithm A G G A C C T A 0 0 0 0 0 2 G 0 0 4 6 0 G 0 10 4 0 A 0 0 2 C 0 0 C 0 T A C C T G G A AGGACCT
Finding pseudoknots anyway • Scores learnt on wild-type sequences • GC different from CG • GC score in stem 1 = #GC in stem 1 / stem 1 length • Accounts for bulges and gaps • Needs threshold to select relevant stems
Typical -1 frameshift site [Brierley, 1989] S2 3’ L1 L’1 S1 L2 5’ AUG NNXXXY YYZ P SP Secondary structure Slippery sequence
Finding pseudoknots anyway 20 nt 50 nt S1.5’ S1.3’ HL from L2 S1.5’ S1.3’ S2.5’ S2.3’
Orpheo • Finds known sites • Fast : 2 minutes on both strands ofS. cerevisiae • Distinguishes 5’ from 3’ and so implicitly accounts for triple interactions • Yields around 200 candidates in yeast (including one with 13% efficiency)
Biological or randomsequences Model Wild-type foldedsequences Score matrix Folding Foldedsequences Mutant foldedsequences Rules Voting Folded and sorted sequences New FS sites In silico andin vivovalidation
Example of a rule if SP length 5 and number of Gs in S1.5’ bottom half 3 and number of Gs in S1.5’ 4and %T in S2.5’ 30 and %C in S2.3’ 75 or %G in S1.5' bottom half 80 and %C in L1 45 orSP length 5 and S1.3' length 6 and %C in S1.3' or SP length 5 and number of Gs in S1.5’ bottom half 3 and %C in S1.3’ 70 and %G in S2.3’ 45 or number of As in S1.5' = 0 and number of As in S2.3' = 0 then %FS 5
Biological or randomsequences Wild-type foldedsequences Score matrix Folding Foldedsequences Mutant foldedsequences Rules Voting Folded and sorted sequences New FS sites In silico andin vivovalidation
Refining the model: Machine learning • To identify relevant properties that characterize FS sites • Disjunctive learning: all sequences do not frameshift for the same reasons [Giedroc etal., 2000](or don’t they ? [Michiels et al. 2001])
Covering and prediction If SP length 5 andnumber of G in S1.5’ bottom half 3 and number of G in S1.5’ 4and %T in S2.5’ 35 and %G in S1.5’ 75 thenFS rate 5% Covering of examples: 70 % Examples predicted in test set: 80 % Counterexamples in test set: 0 %
STOP-1 START0 STOP0 0 phase ORF 1 -1 phase ORF 2 Search for protein patterns • Goal: to find new frameshift sites outside the known consensus known proteic patterns
Validation on random sequences • Hypothesis : biologically relevant sequences have been selected and thus are not random • If something is relevant, it is apart from the means
Experimental results published in Bioinformatics • A COMPLETER