Download
1 / 114
1.16k likes | 1.58k Views
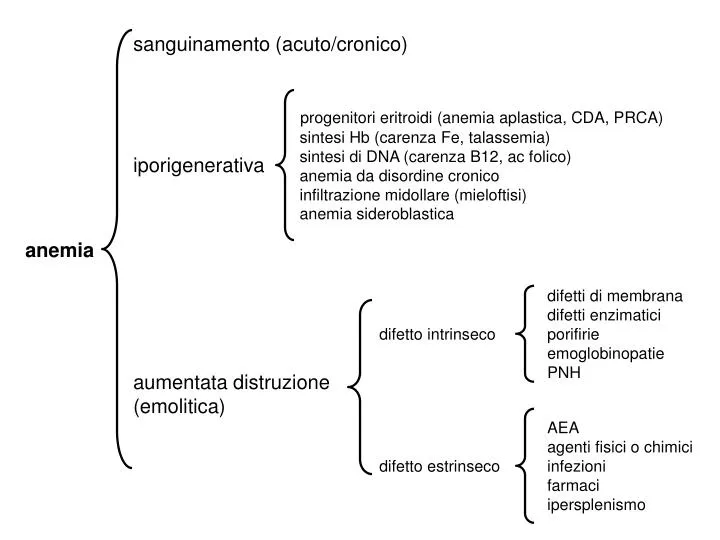
sanguinamento (acuto/cronico). progenitori eritroidi (anemia aplastica, CDA, PRCA) sintesi Hb (carenza Fe, talassemia) sintesi di DNA (carenza B12, ac folico) anemia da disordine cronico infiltrazione midollare (mieloftisi) anemia sideroblastica. iporigenerativa. anemia.

E N D
sanguinamento (acuto/cronico) progenitori eritroidi (anemia aplastica, CDA, PRCA) sintesi Hb (carenza Fe, talassemia) sintesi di DNA (carenza B12, ac folico) anemia da disordine cronico infiltrazione midollare (mieloftisi) anemia sideroblastica iporigenerativa anemia difetti di membrana difetti enzimatici porifirie emoglobinopatie PNH difetto intrinseco aumentata distruzione (emolitica) AEA agenti fisici o chimici infezioni farmaci ipersplenismo difetto estrinseco
THE RED CELL I The simplest cell in the body (no nucleus, no mitochondria, no RNA) Very few kind of proteins (no HLA, no oxydative metabolism) Very modest interactivity Very modest immunogenicity (no HLA class I)
LUNG O2 CO2 O2 O2 CO2 O2 CO2 HCO3- + H+ Arterial blood More Oxyemoglobin less H+, Higher Ph Venous blood Less Oxyemoglobin more H+ Lower Ph HCO3- + H+ O2 CO2 O2 O2 CO2 TISSUE CO2 O2
CAUSES OF ANEMIA IMPAIRED PRODUCTION ACUTE BLOOD LOSS INCREASED DESTRUCTION Hemolytic anemia Hyporegenerative anemia internal external Stem cell is OK but some problems occur during differentiation Of a sick RBC Of healthy blood cells (due to an external problem) Stem cell is damaged (not a true RBC disorder)
HEMOLYTIC ANEMIAS OF HEALTHY BLOOD CELLS Estrinsic causes (usually acquired) Induced by parasites Immunologically mediated Induced by toxins Mechanical cause Autoimmune hemolytic anemia/ transfusion reaction Mechanical valves/ microcirculation disorders Endotoxins/ snake venom malaria
HEMOLYTIC ANEMIAS OF SICK BLOOD CELLS Intrinsic causes (usually inherited) Quantitative Hemoglobin Defect Cytoskeleton/ membrane defect Qualitative Hemoglobin defect RBC metabolism defect GAPDH Deficiency Thalassemia (esp alpha) Spherocytosis Hemoglobinopathies
CONSEQUENCES OF ANEMIA LACK OF O2 IN TISSUES AND COMPENSATION EFFORTS BY THE ORGANISM IN ANY ANEMIA INCREASED PRODUCTION/EXCRETION OF-RBC ASSOCIATED CATABOLYTES IN CASE OF INEFFECTIVE ERYTROPOIESIS OR HEMOLYSIS IN CASE OF ACUTE INTRA-VASCULAR HEMOLYSIS ACUTE HEMOLYTIC SYNDROME
Metabolic enzymes (energy and detoxyfication) Cell membrane (external interactions and transport) Cytoskeleton (structure) GAS-transporting system RED CELL STRUCTURE ANY OF THESE STRUCTURES CAN BE DAMAGED BY SPECIFIC DISORDERS: (IMMUNE HEMOLYTIC ANEMIA SPHEROCYTOSIS, GAPDH DEFICIENCY, HEMOGLOBINOPATHIES)
Dissociazione ossigeno-emoglobina: normale curva sigmoide che correla la saturazione di emoglobina alla parziale pressione di ossigeno (PaO2) a cui è esposta. La curva è spostata a sinistra (è rilasciato meno ossigeno a ogni data PaO2) da un decremento del 2,3 DPG, da un aumento del pH (effetto Bohr) e se l'HbA è sostituita dall'HbF o da un'emoglobina a più alta affinità. La curva è spostata a destra da un aumento del 2,3 DPG attaccato alla Hb, da un decremento del pH e se l'HbA è sostituita dalla HbS oda una HbM (in cui il ferro emico è stabilizzato nella forma ferrica) o se l'Hb è ossidata a metaemoglobina.
HEMOGLOBIN FUNCTION THE O2 DISSOCIATION CURVE
HEMOGLOBIN FUNCTION THE O2 DISSOCIATION CURVE Myoglobin Hb monomer Low 2,3DPG High 2,3DPG Low Ph High Ph High pCO2 Low pCO2 HbF Presence of CO
GAS-TRANSPORTING SYSTEMDISORDERS HEMOGLOBIN DISORDERS HEME DISORDERS (porphyrias, sideropenic anemia CO-intoxication) GLOBIN DISORDERS (Thalassemia, structural hemoglobinopathies)
GLOBIN-DISORDERS QUALITATIVE DISORDERS (structural hemoglobinopathies) QUANTITATIVE DISORDERS (thalassemia) -CHAIN -CHAIN SOLUBILITY STABILITY Oxygen AFFINITY
qualitatively impair the gas trasporting system quantitatively impair the gas transporting system Induce erythroblast or RBC toxicity reduce RBC circulatory performance Hb with altered Hb affinity Thalassemia Sickle-cell disease HB DISEASES CAN CAUSE MANY PROBLEMS
Diseases related to an impaired Hb synthesis Structural Hb disorders associated to reduced Hb synthesis an Structural Hb disorders Acquired Hb disorders Persistence of HbF Sickle-cell disease CO-intoxication thalassemia Hb Lepore GAS-TRANSPORTING SYSTEM DISORDERS HEMOGLOBIN DISORDERS
GAS-TRANSPORTING SYSTEMDISORDERS HEMOGLOBIN DISORDERS HEME DISORDERS (porphyrias, sideropenic anemia CO-intoxication) GLOBIN DISORDERS (Thalassemia, structural hemoglobinopathies)
GLOBIN-DISORDERS QUALITATIVE DISORDERS (structural hemoglobinopathies) QUANTITATIVE DISORDERS (thalassemia) -CHAIN -CHAIN SOLUBILITY STABILITY Oxygen AFFINITY
In some areas there is a clear survival avantage for the preservation of these genes. This is usually associated to a survival advantage for heterozygotes
Disease severity Disease severity observed INCIDENCE SOCIAL RELEVANCE expected Inherited Hb defects are the most common mendelian genetic disorders: 200,000,000 people for thalassemia CLINICAL SYMPTOMS RANGE FROM SILENTTO LETHAL IN UTERO
The human-anopheles-plasmodium ecosystem
Classificazione dell’emoglobinopatie e talassemie: alterazioni strutturali dell’Hb: • anomala polimerizzazione Hb (HbS) • anomala cristallizzazione Hb (HbC) • emoglobine instabili • emoglobine con affinita’ per l’O2 (policitemia); • emoglobine con ¯ affinita’ per l’O2 (cianosi); difetti quantitativi nella produzione delle catene globiniche • a-talassemia • b-talassemia • db-talassemia, gdb-talassemia, ab-talasemia persistenza di Hb fetale (HbF) • pancellulare • eterocellulare emoglobinopatie acquisite • meta-emoglobinemia • sulfo-emoglobinemia • carbossi-emoglobinemia • incremento HbF (chemioterapia, ripresa midollare, mielodisplasie)
DIAGNOSI -TALASSEMIA ETEROZIGOTE Sospetto per familiarità e/o microcitemia Lieve anemia (Hb 9,5 – 11,5 g/dl) con GR aumentati (5- 6,5 x 106/l), riduzione MCV (60-75 /l) ed MCH. Controllare sideremia: se normale eseguire elettroforesi Hb: diagnosi se HbA2 >3-3,5%. Se sideropenia, normalizzare sideremia prima di elettroforesi.
-TALASSEMIA OMOZIGOTE Gravità dell’ anemia dipende da: eventuale produzione residua di catene : genotipo 0/0: malattia più grave, produzione solo di HbF e HbA2 genotipo +/ +:talassemia intermedia quantità di Hb F e HbA2 prodotta: malattia molto meno grave se persistenza HbF (es. talassemie con ampie delezioni)
TERAPIA -TALASSEMIA OMOZIGOTE TERAPIA DI SUPPORTO Trasfusioni per mantenere Hb > 10 g/dl: normale crescita e prevenzione deformità scheletriche. Terapia chelante del ferro con desferrioxamina per infusione s.c. giornaliera di 8- 12 ore. In fase di sperimentazione chelanti orali. SOPRAVVIVENZA MEDIANA 35-40 anni. TRAPIANTO DI MIDOLLO ALLOGENICO Se donatore HLA identico e trapianto entro i 10 anni: 80% guarigione definitiva; 10% mortalità.
TERAPIA -TALASSEMIA OMOZIGOTE APPROCCI SPERIMENTALI Tentativi di ripristinare la sintesi di HbF mediante riattivazione dei genicon agenti de-metilanti del DNA (5-aza-citidina) e inibitori dell’ istone – deacetilasi (butirrati e nuove molecole).
GLOBIN-DISORDERS QUALITATIVE DISORDERS (structural hemoglobinopathies) QUANTITATIVE DISORDERS (thalassemia) -CHAIN -CHAIN SOLUBILITY STABILITY Oxygen AFFINITY
External AA Internal AA Heme pocket AA HB DEFECTS OFTEN ARE OFTEN ASSOCIATED TO POINT MUTATIONS SOLUBILITY STABILITY Oxygen AFFINITY
The most common qualitative Hb disease affects Hb solubility A PROTOTYPE OF A POINT MUTATION DISEASE 6 Glu--> Val SICKLE CELL DISEASE (HBS) NB 6 Glu-->Lys give rise to HBC with a milder phenotype
THE SAME MUTATION AROSE IN DIFFERENT SUBJECTS AND WAS POSITIVELY SELECTED
THE -6 mutation affects Hb sulubility mostly in the deoxygenated state