Download

1 / 38
380 likes | 399 Views
In-depth theoretical analysis of electron behavior in Fe8 complex, showcasing intricate spin interactions using advanced computational methods. Discusses challenges in determining ground states and interpreting spin functions accurately.

E N D
Theoretical description of electrons in single molecule magnets Ernest R Davidson Universities of Washington and North Carolina
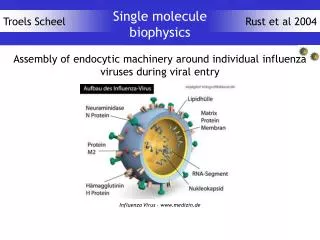
[Fe8(C6H12N3H3)6(OH)12O2]+8 S=10 Note μ3 O's FeIII(d5,S=5/2) 40 singly occupied orbitals
CASSCF or CASCI not feasible 40 singly occupied orbitals coupled as S=10 ~240 Slater determinants 847,660,528 with M=10 574,221,648 combinations with S=10, M=10 6,328 combinations with eight S=5/2 atoms coupled to give S=10, M=10 Would need to compute all S to show 10 was ground state
Note assumed one-to-one correspondence between Heisenberg spin functions and Slater determinants: αAαAβBβB Det{(closed shells of A)φA1αφA2α (closed shells of B) φB1βφB2β}
Reminder: Defines S Defines M This seems to be major source of confusion.
Similarly a pair of d5 S = 5/2 centers like Mn…Mn αAαAαAαAαA βBβBβBβBβB is 0.4% S=5, 4% S=4, 14% S=3, 30% S=2, 36% S=1, and only 17% S=0 even though it is 100% M=0
[Fe8(C6H12N3H3)6(OH)12O2]+8 S=10 Picture shows M = 10 NOT S=10!
C. Cañada PhD Thesis 2002 S = 5 6 FeIII(d5,S=5/2) 30 singly occupied orbitals
Picture shows lowest energy spin orientation, not S2 eigenfunction! H=E0-2∑JABSA•SB ZINDO calculation
Noodleman and Yamaguchi: For two magnetic centers, calculate spin-unrestricted energies and Ŝ2for SA ≥ SB, MA = SA, MB = ±SB, so M = |SA±SB| with values for SA and SB assumed. Substitute in averaged Heisenberg Hamiltonian H = E0– 2J ŜA•ŜB Ŝ = ŜA + ŜB 2 ŜA•ŜB = Ŝ2 – SA(SA+1) – SB(SB+1) and solve for J and E0. Use calculated value or M2+SA+SB for Ŝ2. Then the correct spin-eigenfunction energy levels are E = E0 – J [S(S+1) – SA(SA+1) – SB(SB+1)] for S= SA – SB to SA + SB in steps of 1.
SA2 in black above atoms SA•SB in black above bonds SzA in blue Ideal S2 = M2 +SA+SB Mn2(OH)4(H2O)2 MnII d5 S=5/2
Ligand separated MnII(d5S=5/2) A prototype dinuclear complex Lowest E for S =0 or 5 E=J/2[S(S+1)-35/2], S = 0,1,2,3,4,5
For more than two spin centers follow Noodleman and Yamaguchi example: Substitute in averaged Heisenberg Hamiltonian H = E0 –ΣJABSA •SB from enough calculations to give E0 and all JAB. Then diagonalize H = E0–Σ JAB SA•SB in direct product basis of atomic spin eigenfunctions to get energy levels.
Deliberately misuse DFT calculations. For Treat Kohn-Sham determinant as though it were a wave function to evaluate with projected operator. Or else use chemical intuition value in agreement with spin-space interpretation of Heisenberg Hamiltonian.
Consider a molecule with 3 high-spin d5 transition metal atoms in a triangle. If all J were equal for d5 S=5/2 atoms in a triangle, H = -2J[ŜA•ŜB + ŜB•ŜC + ŜA•ŜC] E = -J[S(S+1) -105/4] S = ½ (2) ; S=3/2 (4) ; S=5/2 (6) ; S=7/2 (5) ; S=9/2 (4) ; S=11/2 (3) ; S=13/2 (2) ; S=15/2 (1) J derived from calculations with αA5αB5αC5 M=15/2 100% S=15/2 αA5αB5βC5 M=5/2 55% S=5/2, 30% S=7/2, 12% S=9/2 …
Mn3O(OH)4(H2O)4 AIM charge
For four or more magnetic centers we begin to get intermediate spin as the ground state. Many states of each spin. Maximum spin is the sum of the SA.
Mn4O2(OH)4(H2O)6 MnII4
Usually SA is assigned by chemical intuition! Unclear how to extract a value for SA from a wave function. J is obtained by least-squares fit to experimental data. Less clear how to obtain J from calculations. Unclear how accurate Heisenberg-Hamiltonian model is.
where either Or else
Basic assumption in extracting J from broken spin computations, DFT or UHF, is that the same energies are just mixed with different weights. This is easily tested for UHF by extracting the ES by spin projection (PUHF). Results show this assumption is NOT valid for UHF! Not testable for DFT because PDFT is not defined. Much debate in the literature about meaning of DFT with broken spin.
Stretched H2 The prototype diradical
H2 aug-cc-pvqz Localized, broken symmetry, broken spin for M=0, R > 1.8
H2 aug-cc-pvqz H2 aug-cc-pvqz
END The standard model is to use the Heisenberg Hamiltonian with parameters from fitting experiment or from DFT or UHF broken spin calculations. There is no check on accuracy of individual states derived in this way. There is evidence that broken spin calculations for different M differ in ESand not just in the weights. There is evidence that assumed site S values have meaning even though site charges do not match assumed oxidation numbers. Broken spin DFT calculations do not give the energy ES with S = M as often assumed for mono-radical and di-radical.