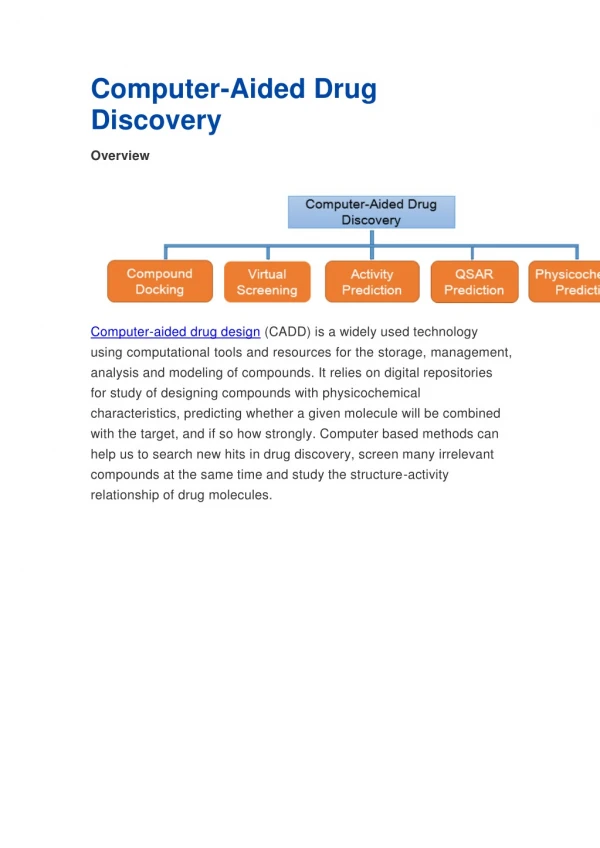
Download

1 / 34
360 likes | 527 Views
Computer simulations in drug design, and the GRID. Dr Jonathan W Essex University of Southampton. Computer simulations?. Molecular dynamics Solve F = ma for particles in system Follow time evolution of system Average system properties over time Monte Carlo Perform random moves on system

E N D
Computer simulations in drug design, and the GRID Dr Jonathan W Essex University of Southampton
Computer simulations? • Molecular dynamics • Solve F = ma for particles in system • Follow time evolution of system • Average system properties over time • Monte Carlo • Perform random moves on system • Accept or reject move on the basis of change in energy • No time information
Free energy simulations Digital filtering Parallel tempering QM/MM Free energy simulations Free energy simulations Structure generation Free energy simulations Modelling Drug action • Administration • Oral, intravenous… • pKa, solubility • Transport and permeation • Passage through membranes • Partition coefficient, permeability • Protein binding • Binding affinity, specificity • Allosteric effects • Metabolism • P450 enzymes • Catalysis
Three main areas of work • Protein-ligand binding • Structure prediction • Binding affinity prediction • Membrane modelling and small molecule permeation • Bioavailability • Coarse-grained membrane models • Protein conformational change • Induced-fit • Large-scale conformational change
Flexible protein docking Most successful structure with experiment (transparent) Most successful structure, experiment, and isoenergetic mode Small molecule docking
Replica-exchange free energy • Free energy methods that are applied between exchanges are the same as normal • Exchanges require little extra computational cost
Z-constraint z Free Energies Membrane permeation z1 z2
Drugs: -blockers alprenolol atenolol pindolol • not “exotic” chemical groups • not too big • similar structure, pKa and weight, but different lipophilicity and oral absorption • much experimental data about Caco-2 permeabilities
Reversible digitally filtered MD • Conformational change associated with low frequency vibrations • Use digital filter applied to atomic velocities to amplify low frequency motion
RDFMD • Effect of a quenching filter
Comb-e-Chem • EPSRC Grid computing project • Main objectives: • Equipment on the Grid – high-throughput small molecule crystallography • E-lab – pervasive computing, provenance • Structure modelling and property prediction • Automatic calculations performed on deposition of a new structure to the database • Distributed (Grid) computing
Distributed computing • Heterogeneous computers – personal PCs • Rather unstable • Network often slow • Suited for embarrassingly parallel calculations • SETI@Home, Folding@Home etc. • E-malaria (JISC) • Poor-man’s resource • Cheap • Large amount of power available
Condor • Condor project started in 1988 to harness the spare cycles of desktop computers • Available as a free download from the University of Wisconsin-Madison • Runs on Linux, Unix and Windows. Unix software can be compiled for Windows using Cygwin • For more information see http://www.cs.wisc.edu/condor/
Distributed computing and replica exchange • Run multiple simulations is parallel • Use different conditions in different simulations to enhance conformational change (e.g. temperature) 300 K 320 K 340 K 360 K
320 K 300 K 340 K 360 K 380 K 400 K
320 K 300 K 340 K 360 K 380 K 400 K 1
320 K 300 K 340 K 360 K 380 K 400 K 1 1 1 1 1 1
320 K 300 K 340 K 360 K 380 K 400 K 2 2 2 2 2 2
320 K 300 K 340 K 360 K 380 K 400 K 3 3 3 2 3 2
320 K 300 K 340 K 360 K 380 K 400 K 4 5 3 2 3 4
320 K 300 K 340 K 360 K 380 K 400 K Catch-up cluster 4 5 3 2 3 4
320 K 300 K 340 K 360 K 380 K 400 K Catch-up cluster 4 5 4 4 4 4
Red spots show when the owner of the node is using it (interrupting our job) Yellow bars show when the job is moved over to the catchup cluster Blue bars represent a node running an even iteration Green bars represent a node running an odd iteration Activity of nodes over the last day
University network failed! Condor master server crashed! Catchup cluster redesignated to six fast, dedicated nodes. Catchup cluster was a large collection of non-dedicated nodes. One slow or interrupted iteration delays large parts of the simulation Activity of nodes since the start of the simulation
Current paradigm for biomolecular simulation • Target selection: literature based; interesting protein/problem • System preparation: highly interactive, slow, idiosyncratic • Simulation: diversity of protocols • Analysis: highly interactive, slow, idiosyncratic • Dissemination: traditional – papers, talks, posters • Archival: archive data… and then lose the tape!
Application Distributed Query 2nd Level Metadata – describing the results of generic analyses Analyse Data Simulation Data 1st Level Metadata – describing the simulation data Distributed Raw Data Managing MD data: BioSimGRID York Nottingham Birmingham Oxford • www.biosimgrid.org • Distributed database environment • Software tools for interrogation and data-mining • Generic analysis tools • Annotation of simulation data Bristol London Southampton
Comparative simulations • Increase significance of results • Effect of force field • Simulation protocol • Long simulations and multiple simulations • Biology emerges from the comparisons • Very easy to over-interpret protein simulations • What’s noise, and what’s systematic?
Test Application: Comparison of Active Site Dynamics • OMPLA – bacterial outer membrane lipase; GROMACS; Oxford • AChE – neurotransmitter degradation at synapses; NWChem; UCSD (courtesy of Andrew McCammon) • Both have catalytic triad at active site – compare conformational dynamics
Traj Query Tool Other SQL Editor AAA Module Video/Img Generator HTML Generator Analysis Tool HTTP(S) TCP/IP TCP/IP DB2 Cluster BioSimGRID prototype WEB Python Environment Web Portal Environment Apache/SSL Python Applications Server Database Access: DBI / PythonDB2 / PortalLib Prototype: Summer 2003 (UK e-science All-Hands meeting)
BioSimGrid Config. Files Hybrid Storage Analysis Toolkit Distances RMSD Simulation Result Files RMSF Internal Angles Volume Surface database Flat file 4. Analysis User input 3. Data- on-demand Query 5. View Result Metadata Visualisation Tools 2. Generation of Metadata Trajectory 1. Submission of trajectory Revised structure
Example of script use (distance matrix) FC = FrameCollection(‘2,5-8’) myDistanceMatrix = DistanceMatrix(FC) myDistanceMatrix.createPNG() myDistanceMatrix.createAGR() • Script Calculates Distance Matrix • User has requested result as PNG • Grace project file was also produced www.biosimgrid.org
Future directions: Multiscale biomolecular simulations QM drug binding protein motions drug diffusion Bristol Southampton Oxford London • Membrane bound enzymes – major drug targets (cf. ibruprofen, anti-depressants, endocannabinoids); gated access to active site coupled to membrane fluctuations • Complex multi-scale problem: QM/MM; ligand binding; membrane/protein fluctuations; diffusive motion of substrates/drugs in multiple phases • Need for integrated simulations on GRID-enabled HPC resources
Linux cluster HPCx Computational challenges IntBioSim BioSimGRID database www.biosimgrid.org • Need to integrate HPC, cluster & database resources • Funding: bid to BBSRC under consideration…
My group: Stephen Phillips Richard Taylor Daniele Bemporad Christopher Woods Robert Gledhill Stuart Murdock My funding: BBSRC, EPSRC, Royal Society, Celltech, AstraZeneca, Aventis, GlaxoSmithKline My collaborators: Mark Sansom, Adrian Mulholland, David Moss, Oliver Smart, Leo Caves, Charlie Laughton, Jeremy Frey, Peter Coveney, Hans Fangohr, Muan Hong Ng, Kaihsu Tai, Bing Wu, Steven Johnston, Mike King, Phil Jewsbury, Claude Luttmann, Colin Edge Acknowledgements