Download

1 / 36
410 likes | 902 Views
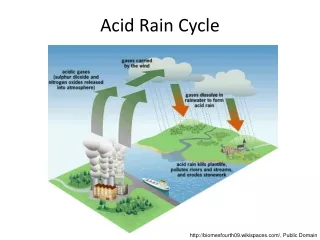
Tricarboxylic Acid Cycle. UNIT II: Intermediary Metabolism. Figure 9.1. The tricarboxylic acid cycle shown as a part of the central pathways of energy metabolism. Overview. TCA cycle (a.k.a Krebs cycle or citric acid cycle) plays several roles in metabolism

E N D
Tricarboxylic Acid Cycle UNIT II: Intermediary Metabolism
Figure 9.1. The tricarboxylic acid cycle shown as a part of the central pathways of energy metabolism.
Overview • TCA cycle (a.k.a Krebs cycle or citric acid cycle) plays several roles in metabolism • It is the final pathway where oxidative metabolism of CHO’s, aa’s & fatty acids converge, their C skeletons being converted to CO2 & H2O. This oxidation provides energy for production of majority of ATP. • The cycle occurs in mitoch & is in close proximity to the reactions of e-transport, which oxidize the reduced coenzymes produced by the cycle • The TCA cycle is thus an aerobic pathway, because O2 is required as final e-acceptor • The cycle also participates in a number of synthetic reactions. E.g., it functions in formation of gluc from C skeletons of some aa’s, & it provides building blocks for synthesis of some aa’s & heme. • Intermediates of TCA cycle can also be synthesized by catabolism of some aa’s. • This cycle should not be viewed as a closed circle, but instead as a traffic circle with cpds entering & leaving as required.
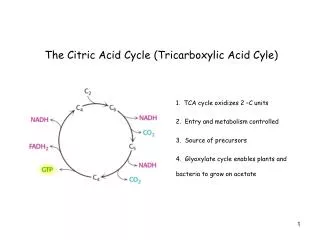
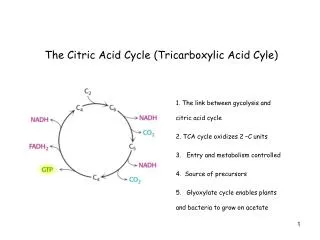
II. Reactions of the TCA cycle • In TCA cycle, OAA is first condensed with an acetyl group from acetyl CoA, & then is regenerated as the cycle is completed. Thus, entry of one acetyl CoA into one round of the cycle does not lead to the net production or consumption of intermediates A. Oxidative decarboxylation of pyruvate • Pyruvate, must be transported to mitoch before it can enter TCA cycle. This is accomplished by a specific pyruvate transporter • Once in matrix, pyruvate is converted to acetyl CoA by pyruvate dehydrogenase complex Note: irreversibility of reaction precludes formation of pyruvate from acetyl CoA, and explains why gluc can’t be formed from acetyl CoA via gluconeogenesis
Figure 9.2 Oxidative decarboxylation of pyruvate.
Strictly speaking, pyruvate dehydrogenase complex is not part of TCA cycle proper, but is a major source of acetyl CoA, the 2C substrate of the cycle 1. Component enzymes: • Pyruvate dehydrogenase complex is a multimolecular aggregate of 3 enz’s: pyruvate dehydrogenase (E1, a.k.a a decarboxylase), dihydrolipoyl transacetylase (E2), & dihydrolipoyl dehydrogenase (E3). • Each is present in multiple copies, and each catalyzes a part of the overall reaction. • Their physical association links the reactions in proper sequence without release of intermediates • In addition to the enzymes participating in conversion of pyruvate to acetyl CoA, the complex also contains 2 tightly bound regulatory enzymes, protein kinase & phosphoprotein phosphatase
Figure 9.3 Mechanism of action of the pyruvate dehydrogenase complex. TPP = thiamine pyrophosphate; L = lipoic acid.
2. Coenzymes: - The pyruvate dehydrogenase complex contains 5 coenzymes that act as carriers or oxidants for the intermediates of the reactions shown in Fig 9-3. • E1 requires thiamine pyrophosphate, E2 requires lipoic acid & coenzyme A, and E3 requires FAD & NAD+ Note: deficiencies of thiamine or niacin can cause serious CNS problems. This is because brain cells are unable to produce sufficient ATP (via TCA cycle) for proper function if pyruvate dehydrogenase is inactive 3. Regulation of pyruvate dehydrogenase complex: - The 2 regulatory enzymes that are part of the complex alternately activate & inactivate E1: the cAMP-independent protein kinase phosphorylates and, thereby, inhibits E1, whereas phosphoprotein phosphatase activates E1
The kinase is allosterically activated by ATP, acetyl CoA, and NADH. Therefore, in presence of these high-energy signals, the pyruvate dehydrogenase complex is turned off • Acetyl CoA and NADH also allosterically inhibit the dephosphorylated (active) form of E1. • Protein kinase is allosterically inactivated by NAD+ and coenzyme A, low energy signals that thus turn pyruvate dehydrogenase on • Pyruvate is also a potent inhibitor of protein kinase. Therefore, if pyruvate conc’s are elevated, E1 will be maximally active • Calcium is a strong activator of protein phosphatase, stimulating E1 activity. Note: this is particularly important in skeletal muscle, where release of Ca2+ during contraction stimulates the pyruvate dehydrogenase complex, and thereby energy production
Figure 9.4 Regulation of pyruvate dehydrogenase complex.
4. Pyruvate dehydrogenase deficiency • A deficiency in the pyruvate dehydrogenase complex is the most common biochemical cause of congenital lactic acidosis • This enz deficiency results in an inability to convert pyruvate to acetyl CoA pyruvate shunted to lactic acid via lactate dehydrogenase • This causes particular problems for the brain, which relies on TCA cycle for most its energy, & is particularly sensitive to acidosis • The most severe form of this deficiency causes overwhelming lactic acidosis with neonatal death • A 2nd form produces moderate lactic acidosis, but causes profound psychomotor retardation, with damage to cerebral cortex, basal ganglia and brain stem death in infancy
A 3rd form causes episodic ataxia (an inability to coordinate voluntary muscles) that is induced by a CHO-rich meal • The E1 defect is X-linked, but because the importance of the enz in the brain, it affects both males & females. Therefore, the defect is classified as X-linked dominant • There is no proven treatment for pyruvate dehydrogenase complex deficiency, although a ketogenic diet (one low in CHO & enriched in fats) has been shown in some cases to be of benefit. Such a diet provides an alternate fuel supply in form of ketone bodies that can be used by most tissues including the brain, but not the liver
5. Mechanism of arsenic poisoning • As previously described, arsenic can interfere with glycolysis at glyceraldehyde-3P step, thereby decreasing ATP production • “Arsenic poisoning” is, however, due primarily to inhibition of enz’s that require lipoic acid as a cofactor, including pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and branched-chain α-keto acid dehydrogenase • Arsenite (the trivalent form of arsenic) forms a stable complex with thiol (-SH) groups of lipoic acid, making that cpd unavailable to serve as a coenzyme • When it binds to lipoic acid in pyruvate dehydrogenase complex, pyruvate (and consequently lactate) accumulate. Like pyruvate dehydrogenase complex deficiency, this particularly affects brain causing neurologic disturbances and death
B. Synthesis of citrate from acetyl CoA and OAA • Condensation of acetyl CoA & OAA to form citrate is catalyzed by citrate synthase. This aldol condensation has an equil far in direction of citrate synthesis • Citrate synthase is allosterically activated by Ca2+ & ADP, & inhibited by ATP, NADH, succinyl CoA, & fatty acyl CoA derivatives • However, primary mode of regulation is also determined by availability of its substrates, acetyl CoA & OAA Note: - Citrate, in addition to being an intermediate in TCA cycle, provides a source of acetyl CoA for cytosolic synthesis of fatty acids - Citrate also inhibits PFK, the rate-setting enz of glycolysis, & activates acetyl CoA carboxylase (the rate-limiting enz of fatty acid synthesis)
Figure 9.5 Formation of a-ketoglutarate from acetyl CoA and oxaloacetate.
C. Isomerization of citrate • Citrate is isomerized to isocitrate by aconitase Note: • Aconitase is inhibited by fluoroacetate, a cpd that is used as a rat poison. Fluoroacetate is converted to fluoroacetyl CoA, which condenses with OAA to form fluorocitrate, a potent inhibitor of aconitase, resulting in citrate accumulation D. Oxidation and decarboxylation of isocitrate - Isocitrate dehydrogenase catalyzes the irreversible oxidative decarboxylation of isocitrate, yielding the 1st of three NADH molecules produced by the cycle, & 1st release of CO2 - This is one of rate-limiting steps of TCA cycle. The enz is allosterically activated by ADP (low energy signal) and Ca2+, and is inhibited by ATP and NADH, whose levels are elevated when cell has abundant energy stores
E. Oxidative decarboxylation of α-KG • Conversion of α-KG to succinyl CoA is catalyzed by the α-KG dehydrogenase complex, which consists of 3 enzymatic activities • The mechanism of this oxidative decarboxylation is very similar to that used for conversion of pyruvate to acetyl CoA • The reaction releases the 2nd CO2 and produces the 2nd NADH of the cycle. • The coenzymes required are thiamine pyrophosphate, lipoic acid, FAD, NAD+, and coenzyme A. each functions as part of the catalytic mechanism in a way analogous to that described for pyruvate dehydrogenase complex
The equil of reaction is far in direction of succinyl CoA, a high-energy thioester similar to acetyl CoA. • α-KG dehydrogenase complex is inhibited by ATP, GTP, NADH, and succinyl CoA, and activated by Ca2+ • However, it is not regulated by phospho/dephospho reactions as described for pyruvate dehydrogenase complex Note: α-KG is also produced by oxidative deamination or transamination the aa glu
F. Cleavage of succinyl CoA • Succinate thiokinase (a.k.a succinyl CoA synthetase) cleaves the high-energy thioester bond of succinyl CoA • This reaction is coupled to phospho of GDP to GTP. GTP and ATP are energetically interconvertible by the nucleoside diphosphate kinase reaction: GTP + ADP ↔ GDP + ATP - Generation of GTP by succinate thiokinase is another e.g. of substrate-level phospho. Note: succinyl CoA is also produced from propionyl CoA derived from metabolism of fatty acids with an odd # of C atoms, & from metabolism of several aa’s (e.g., ile, val)
G. Oxidation of succinate • Succinate is oxidized to fumarate by succinate dehydrogenase, producing reduced coenzyme FADH2 (FAD rather than NAD+, is the e-acceptor because the reducing power of succinate is not sufficient to reduce NAD+) • Succinate dehydrogenase is inhibited by OAA H. Hydration of fumarate • Fumarate is hydrated to malate in a freely reversible reaction catalyzed by fumarase (= fumarate hydratase) Note: fumarate is also produced by urea cycle, in purine synthesis, and during catabolism of the aa’s, phe & tyr.
I. Oxidation of malate • Malate is oxidized to OAA by malate dehydrogenase. This reaction produces 3rd and final NADH of the cycle. Note: OAA is also produced by transamination of the aa, Asp.
III. Energy produced by the TCA cycle • Two C atoms enter the cycle as acetyl CoA & leave as CO2. • The cycle does not involve net consumption or production of OAA or any other intermediate • Four pairs of e’s are transferred during one turn of the cycle: 3 pairs of e’s reducing NAD+ to NADH & one reducing FAD to FADH2. • Oxidation of one NADH by ETC leads to formation of ~ 3 ATP, whereas oxidation of FADH2 yields ~ 2 ATP • Total yield of ATP from oxidation of one acetyl CoA is:
Figure 9.8. Number of ATP molecules produced from the oxidation of one molecule of acetyl CoA (using both substrate-level and oxidative phosphorylation).
IV. Regulation of the TCA cycle A. Regulation by activation and inhibition of enzyme activity • In contrast to glycolysis which is regulated primarily by PFK, the TCA cycle is controlled by regulation of several enz activities. The most important of these are citrate synthase, isocitrate dehydrogenase, & α-KG dehydrogenase complex
B. Regulation by availability of ADP 1. Effect of elevated ADP: - Energy consumption as a result of muscular contraction, biosynthetic reactions or other processes result in hydrolysis of ATP to ADP & Pi. • Resulting increase in conc of ADP accelerates rate of reactions that use ADP to generate ATP, most important of which is oxphos • Production of ATP increases until it matches rate of ATP consumption by energy-requiring reactions
2. Effect of low ADP: • If ADP (or Pi) is present in limiting conc, formation of ATP by oxphos decreases as a result of the lack of phosphate acceptor (ADP) or inorganic phosphate (Pi) • The rate of oxphos is proportional to [ADP][Pi]/[ATP]; this is known as “respiratory control of energy production” • Oxidation of NADH & FADH2 by ETC also stops if ADP is limiting. This is because the processes of oxidation & phospho are tightly coupled & occur simultaneously • As NADH & FADH2 accumulate, their oxidized forms become depleted causing oxidation of acetyl CoA by the TCA cycle to be inhibited as a result of a lack of oxidized coenzymes
Figure 9.9. A. production of reduced coenzymes, ATP, and CO2 in TCA cycle. B. inhibitors and activators of the cycle.
Summary • Pyruvate is oxidatively decarboxylated by pyruvate dehydrogenase complex producing acetyl CoA, which is the major fuel for TCA cycle • This enz complex requires five coenzymes: thiamine pyrophosphate, lipoic acid, FAD, NAD+, and coenzyme-A (which contains the vitamin pantothenic acid) • The reaction is activated by NAD+, coenzyme-A, and pyruvate, and inhibited by ATP, acetyl CoA, NADH, and Ca2+. • Pyruvate dehydrogenase deficiency is the most common biochemical cause of congenital lactic acidosis. Because the deficiency deprives the brain of acetyl CoA, the CNS is particularly affected, with profound psychomotor retardation & death occurring in most patients. The deficiency is X-linked dominant • Arsenic poisoning causes inactivation of pyruvate dehydrogenase by binding to lipoic acid
Citrate is synthesized fro OAA and acetyl CoA by citrate synthase. This enz is allosterically activated by ADP, and inhibited by ATP, NADH, succinyl CoA, and fatty acyl CoA derivatives • Citrate is isomerized to isocitrate by aconitase. Isocitrate is oxidized & decarboxylated by isocitrate dehydrogenase to α-KG, producing CO2 and NADH. The enz is inhibited by ATP & NADH, & is activated by ADP & Ca2+. • α-KG is oxidatively decarboxylated to succinyl CoA by α-KG dehydrogenase complex, producing CO2 & NADH. The enz is very similar to pyruvate dehydrogenase and uses the same coenzymes. • α-KG dehydrogenase complex is activated by Ca2+, and inhibited by ATP, GTP, NADH, & succinyl CoA.
Succinyl CoA is cleaved by succinate thiokinase (= succinyl CoA synthetase), producing succinate and GTP. This is an e.g. of substrate-level phospho. • Succinate is oxidized to fumarate by succinate dehydrogenase, producing FADH2. this enz is inhibited by OAA. • Fumarate is hydrated to malate by fumarase (= fumarate hydratase), and malate is oxidized to OAA by malate dehydrogenase, producing NADH. • Three NADH, one FADH2, and one GTP (whose terminal phosphate can be transferred to ADP by nucleoside diphosphate kinase, ATP) are produced by one round of TCA cycle. • Oxidation of NADHs and FADH2 by ETC yields ~ 11 ATPs, making 12 the total # of ATPs produced
Figure 9.10 Key concept map for tricarboxylic acid cycle.
















![Tricarboxylic acid cycle (TCA Cycle) [Kreb’s cycle] [Citric acid cycle]](https://cdn3.slideserve.com/6696193/tricarboxylic-acid-cycle-tca-cycle-kreb-s-cycle-citric-acid-cycle-dt.jpg)