Download
1 / 1
10 likes | 141 Views
Computational tools to aid identification of potential horizontally transferred genes involved in pathogenicity

E N D
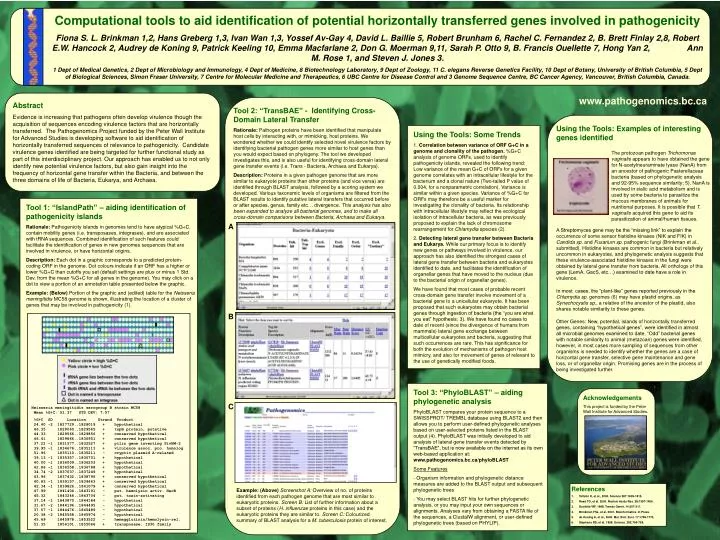
Computational tools to aid identification of potential horizontally transferred genes involved in pathogenicity Fiona S. L. Brinkman 1,2, Hans Greberg 1,3, Ivan Wan 1,3, Yossef Av-Gay 4, David L. Baillie 5, Robert Brunham 6, Rachel C. Fernandez 2, B. Brett Finlay 2,8, Robert E.W. Hancock 2, Audrey de Koning 9, Patrick Keeling 10, Emma Macfarlane 2, Don G. Moerman 9,11, Sarah P. Otto 9, B. Francis Ouellette 7, Hong Yan 2, Ann M. Rose 1, and Steven J. Jones 3. 1 Dept of Medical Genetics, 2 Dept of Microbiology and Immunology, 4 Dept of Medicine, 8 Biotechnology Laboratory, 9 Dept of Zoology, 11 C. elegans Reverse Genetics Facility, 10 Dept of Botany, University of British Columbia, 5 Dept of Biological Sciences, Simon Fraser University, 7 Centre for Molecular Medicine and Therapeutics, 6 UBC Centre for Disease Control and 3 Genome Sequence Centre, BC Cancer Agency, Vancouver, British Columbia, Canada. Abstract Evidence is increasing that pathogens often develop virulence though the acquisition of sequences encoding virulence factors that are horizontally transferred. The Pathogenomics Project funded by the Peter Wall Institute for Advanced Studies is developing software to aid identification of horizontally transferred sequences of relevance to pathogenicity. Candidate virulence genes identified are being targeted for further functional study as part of this interdisciplinary project. Our approach has enabled us to not only identify new potential virulence factors, but also gain insight into the frequency of horizontal gene transfer within the Bacteria, and between the three domains of life of Bacteria, Eukarya, and Archaea. www.pathogenomics.bc.ca Tool 2: “TransBAE” - Identifying Cross-Domain Lateral Transfer Rationale: Pathogen proteins have been identified that manipulate host cells by interacting with, or mimicking, host proteins. We wondered whether we could identify selected novel virulence factors by identifying bacterial pathogen genes more similar to host genes than you would expect based on phylogeny. The tool we developed investigates this, and is also useful for identifying cross-domain lateral gene transfer events (i.e. Trans - Bacteria, Archaea and Eukarya). Description: Proteins in a given pathogen genome that are more similar to eukaryote proteins than other proteins (and vice versa) are identified through BLAST analysis, followed by a scoring system we developed. Various taxonomic levels of organisms are filtered from the BLAST results to identify putative lateral transfers that occurred before or after species, genus, family etc… divergence. This analysis has also been expanded to analyze all bacterial genomes, and to make all cross-domain comparisons between Bacteria, Archaea and Eukarya. Using the Tools: Examples of interesting genes identified A Streptomyces gene may be the “missing link” to explain the occurrence of some sensor histidine kinases (NIK and FIK) in Candida sp. and Fusarium sp. pathogenic fungi (Brinkman et al., submitted). Histidine kinases are common in bacteria but relatively uncommon in eukaryotes, and phylogenetic analysis suggests that these virulence-associated histidine kinases in the fungi were obtained by lateral gene transfer from bacteria. All orthologs of this gene (LemA, GacS, etc…) examined to date have a role in virulence. In most cases, the “plant-like” genes reported previously in the Chlamydia sp. genomes (6) may have plastid origins, as Synechocystis sp., a relative of the ancestor of the plastid, also shares notable similarity to these genes. Other Genes: New, potential, islands of horizontally transferred genes, containing “hypothetical genes”, were identified in almost all microbial genomes examined to date. “Odd” bacterial genes with notable similarity to animal (metazoan) genes were identified, however, in most cases more sampling of sequences from other organisms is needed to identify whether the genes are a case of horizontal gene transfer, selective gene maintenance and gene loss, or of organellar origin. Promising genes are in the process of being investigated further. Using the Tools: Some Trends 1. Correlation between variance of ORF G+C in a genome and clonality of the pathogen. %G+C analysis of genome ORFs, used to identify pathogenicity islands, revealed the following trend: Low variance of the mean G+C of ORFs for a given genome correlates with an intracellular lifestyle for the bacterium and a clonal nature (Two-tailed P value of 0.004, for a nonparametric correlation). Variance is similar within a given species. Variance of %G+C for ORFs may therefore be a useful marker for investigating the clonality of bacteria. Its relationship with intracellular lifestyle may reflect the ecological isolation of intracellular bacteria, as was previously proposed to explain the lack of chromosome rearrangement for Chlamydia species (2). 2. Detecting lateral gene transfer between Bacteria and Eukarya. While our primary focus is to identify new genes or pathways involved in virulence, our approach has also identified the strongest cases of lateral gene transfer between bacteria and eukaryotes identified to date, and facilitates the identification of organellar genes that have moved to the nucleus (due to the bacterial origin of organellar genes). We have found that most cases of probable recent cross-domain gene transfer involve movement of a bacterial gene to a unicellular eukaryote. It has been proposed that such eukaryotes may obtain bacterial genes through ingestion of bacteria (the “you are what you eat” hypothesis; 3). We have found no cases to date of recent (since the divergence of humans from mammals) lateral gene exchange between multicellular eukaryotes and bacteria, suggesting that such occurrences are rare. This has significance for both the evolution of mechanisms of pathogen host mimicry, and also for movement of genes of relevant to the use of genetically modified foods. The protozoan pathogen Trichomonas vaginalis appears to have obtained the gene for N-acetylneuraminate lyase (NanA) from an ancestor of pathogenic Pasterellaceae bacteria (based on phylogenetic analysis and 92-95% sequence similarity; 5). NanA is involved in sialic acid metabolism and is used by some bacteria to parasitize the mucous membranes of animals for nutritional purposes. It is possible that T. vaginalis acquired this gene to aid its parasitization of animal/human tissues. Tool 1: “IslandPath” – aiding identification of pathogenicity islands Rationale: Pathogenicity islands in genomes tend to have atypical %G+C, contain mobility genes (i.e. transposases, integrases), and are associated with tRNA sequences. Combined identification of such features could facilitate the identification of genes in new genomes sequences that are involved in virulence, or have horizontal origins. Description: Each dot in a graphic corresponds to a predicted protein-coding ORF in the genome. Dot colours indicate if an ORF has a higher or lower %G+C than cutoffs you set (default settings are plus or minus 1 Std. Dev. from the mean %G+C for all genes in the genome). You may click on a dot to view a portion of an annotation table presented below the graphic. Example: (Below) Portion of the graphic and (edited) table for the Neisseria meningitidis MC58 genome is shown, illustrating the location of a cluster of genes that may be involved in pathogenicity (1). A B • Tool 3: “PhyloBLAST” – aiding phylogenetic analysis • PhyloBLAST compares your protein sequence to a SWISSPROT/ TREMBL database using BLAST2 and then allows you to perform user-defined phylogenetic analyses based on user-selected proteins listed in the BLAST output (4). PhyloBLAST was initially developed to aid analysis of lateral gene transfer events detected by “TransBAE”, but is now available on the internet as its own web-based application at: www.pathogenomics.bc.ca/phyloBLAST • Some Features • Organism information and phylogenetic distance measures are added to the BLAST output and subsequent phylogenetic trees • - You may select BLAST hits for further phylogenetic analysis, or you may input your own sequences or alignments. Analyses vary from obtaining a FASTA file of the sequences, a ClustalW alignment, or user-defined phylogenetic trees (based on PHYLIP). Acknowledgements This project is funded by the Peter Wall Institute for Advanced Studies. C Neisseria meningitidis serogroup B strain MC58 Mean %G+C: 51.37 STD DEV: 7.57 %G+C SD Location Strand Product 24.40 -2 1827729..1828019 + hypothetical 46.35 1828060..1829565 + tspB protein, putative 44.33 1829566..1829856 + conserved hypothetical 46.41 1829866..1830951 + conserved hypothetical 37.22 -1 1831577..1832527 + pilin gene inverting PivNM-2 39.95 -1 1834676..1835113 + virulence assoc. pro. homolog 51.96 1835110..1835211 - cryptic plasmid A-related 39.13 -1 1835357..1835701 + hypothetical 40.00 -1 1836009..1836203 + hypothetical 42.86 -1 1836558..1836788 + hypothetical 34.74 -2 1837037..1837249 + hypothetical 43.96 1837432..1838796 + conserved hypothetical 40.83 -1 1839157..1839663 + conserved hypothetical 42.34 -1 1839826..1841079 + conserved hypothetical 47.99 1841404..1843191 - put. hemolysin activ. HecB 45.32 1843246..1843704 - put. toxin-activating 37.14 -1 1843870..1844184 - hypothetical 31.67 -2 1844196..1844495 - hypothetical 37.57 -1 1844476..1845489 - hypothetical 20.38 -2 1845558..1845974 - hypothetical 45.69 1845978..1853522 - hemagglutinin/hemolysin-rel. 51.35 1854101..1855066 + transposase, IS30 family • References • Tettelin H, et al., 2000. Science 287:1809-1815. • Read TD, et al. 2000. Nucleic Acids Res. 28:1397-1406. • Doolittle WF. 1998. Trends Genet. 14:307-311. • Brinkman FSL, et al. 2001. Bioinformatics. In Press. • de Koning A, et al., 2000. Mol. Biol. Evol. 17:1769-1773. • Stephens RS, et al. 1998. Science. 282:754-759. Example: (Above)Screenshot A: Overview of no. of proteins identified from each pathogen genome that are most similar to eukaryotic proteins. Screen B: List of further information about a subset of proteins (H. influenzae proteins in this case) and the eukaryotic proteins they are similar to. Screen C: Colourized summary of BLAST analysis for a M. tuberculosis protein of interest.