Download
1 / 11
110 likes | 277 Views
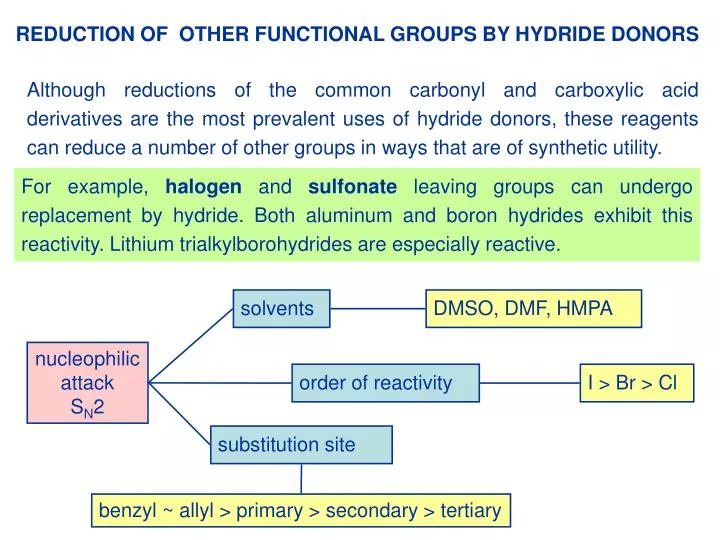
solvents. DMSO, DMF, HMPA. order of reactivity. I > Br > Cl. substitution site. benzyl ~ allyl > primary > secondary > tertiary. REDUCTION OF OTHER FUNCTIONAL GROUPS BY HYDRIDE DONORS.

E N D
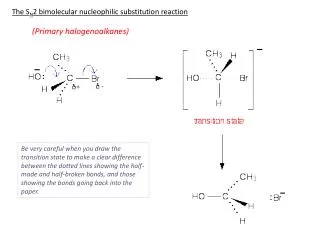
solvents DMSO, DMF, HMPA order of reactivity I > Br > Cl substitution site benzyl ~ allyl > primary > secondary > tertiary REDUCTION OF OTHER FUNCTIONAL GROUPS BY HYDRIDE DONORS Although reductions of the common carbonyl and carboxylic acid derivatives are the most prevalent uses of hydride donors, these reagents can reduce a number of other groups in ways that are of synthetic utility. For example, halogen and sulfonate leaving groups can undergo replacement by hydride. Both aluminum and boron hydrides exhibit this reactivity. Lithium trialkylborohydrides are especially reactive. nucleophilic attack SN2
However, the range of halides that can be reduced includes aryl halides and bridgehead halides, which cannot react by the SN2 mechanism. There is loss of stereochemical integrity in the reduction of vinyl halides, suggesting the involvement of radical intermediates. Formation and subsequent dissociation of a radical anion by one-electron transfer is a likely mechanism for reductive dehalogenation of compounds that cannot react by an SN2 mechanism.
One experimental test for the involvement of radical intermediates is to study 5-hexenyl systems and look for the characteristic cyclization to cyclopentane derivatives. • When 5-hexenyl bromide or iodide reacts with LiAlH4, no cyclization products are observed. However, the more hindered 2,2-dimethyl-5-hexenyl iodide gives mainly cyclic product. The presence of transition-metal ions has a catalytic effect on reduction of halides and tosylates by LiAIH4. Various "copper hydride" reducing agents are effective for removal of halide and tosylate groups. The primary synthetic value of these reductions is for the removal of a hydroxyl function after conversion to a halide or tosylate.
Epoxides are converted to alcohols by LiAlH4. The reaction occurs by nucleophilic attack, and hydride addition at the less hindered carbon of the epoxide is usually observed. Cyclohexene epoxides are preferentially reduced by an axial approach of the nucleophile.
Alkynes are reduced to E-alkenes by LiAlH4 This stereochemistry is complementary to that of partial hydrogenation, which gives Z-isomers. Alkyne reduction by LiAlH4, is greatly accelerated by a nearby hydroxyl group. Typically, propargylic alcohols react in ether or THF over a period of several hours, whereas forcing conditions are required for isolated triple bonds. This is presumably the result of coordination of the hydroxyl group at aluminum and formation of cyclic intermediate. The involvement of intramolecular Al-H addition has been demonstrated by use of LiAlD4, as the reductant. When reduction by LiAlD4 is followed by quenching with normal water, propargylic alcohol gives 3-2H-prop-2-enol. Quenching with D2O gives 2-2H-3-2H-prop-2-enol, indicating overall anti addition.
● Alcohols that can be ionized in trifluoroacetic acid are reduced to hydrocarbons in the presence of a silane. ● Aromatic aldehydes and ketones are reduced to alkylaromatics 5.3. Group IV Hydride Donors Both Si-H and C-H bond containing compounds can function as hydride donors under certain circumstances. The silicon-hydrogen bond is capable of transferring a hydride to carbocations.
● Aliphatic ketones can be reduced to hydrocarbons by triethylsilane and gaseous BF3, which is a sufficiently strong Lewis acid to promote formation of a carbocation from the intermediate alcohol. ● Carboxylic acids and aromatic esters can be reduced to methyl groups in a reaction with HSiCl3 as reducing agent.
There is also a group of reactions in which hydride is transferred from carbon. • The carbon-hydrogen bond has little intrinsic tendency to act as a hydride donor, so especially favorable circumstances are required to observe this reactivity. • Frequently, these reactions proceed through a cyclic transition state in which a new C-H bond is formed simultaneously with the C-H cleavage. • Hydride transfer is facilitated by high electron density at the carbon atom.
Aluminum alkoxides catalyze transfer of hydride from an alcohol to a ketone. This is generally an equlibrium process, and the reaction can be driven to completion if the ketone is removed from the system by distillation, for example. This process is called the Meerwein-Pondorf-Verley reduction. The reaction proceeds via a cyclic transition state involving coordination of both the alcohol and ketone oxygens to the aluminum. Hydride donation usually takes place from the less hindered face of the carbonyl group.
Certain lanthanide alkoxides, such as ButOSmI2, have also been found to catalyze hydride exchange between alcohols and ketones. Isopropanol can serve as the reducing agent for aldehydes and ketones that are thermodynamically better hydride acceptors than acetone. Like the Meenvein-Pondorff-Verley reduction, these reactions are believed to proceed under thermodynamic control, and the more stable stereoisomer is the main product. Another reduction process, catalysed by iridium chloride and characterized by very high axial/equatorial product ratios for cyclohexanones, apparently involves hydride transfer from isopropanol.
Formic acid can also act as a donor of hydrogen. The driving force in this case is the formation of carbon dioxide. • A useful application is the Clark-Eschweiler reductive alkylation of amines. • Heating a primary or secondary amine with formaldehyde and formic acid results in complete methylation to the tertiary amine • The hydride acceptor is the iminium ion resulting from condensation of the amine with formaldehyde.

![Transannular Cyclization of Dehydrobenzo [12] annulene Induced by Nucleophilic Attack](https://cdn1.slideserve.com/1590357/transannular-cyclization-of-dehydrobenzo-12-annulene-induced-by-nucleophilic-attack-dt.jpg)